Command Palette
Search for a command to run...
L'équipe De l'Université Westlake a Ouvert Le Code Source De SaProt Et d'autres Modèles De Langage Protéique, Couvrant La Prédiction De La Fonction De Structure, La Recherche d'informations Intermodales, La Conception De Séquences d'acides Aminés, etc.

Les 22 et 23 mars 2025, le « AI Protein Design Summit » de l'Université Jiao Tong de Shanghai s'est officiellement tenu.Le sommet a réuni plus de 300 experts et universitaires d'universités renommées telles que l'Université Tsinghua, l'Université de Pékin, l'Université Fudan, l'Université du Zhejiang et l'Université de Xiamen, ainsi que plus de 200 représentants d'entreprises industrielles de premier plan et de personnel technique de R&D, pour discuter en profondeur des derniers résultats de recherche, des avancées technologiques et des perspectives d'application industrielle de l'IA dans le domaine de la conception de protéines.

Lors du sommet,Le Dr Yuan Fajie de l'Université Westlake a partagé les derniers progrès de la recherche sur les modèles de langage protéique sur le thème « Recherche et application des modèles de langage protéique » et a présenté en détail les réalisations importantes de l'équipe.Y compris les modèles de langage protéique SaProt, ProTrek, Pinal, Evolla, etc. HyperAI a organisé et résumé le partage approfondi sans violer l'intention initiale. Voici une transcription des points saillants du discours.
Un modèle de langage protéique à noter
Les protéines sont des macromolécules biologiques composées de 20 acides aminés liés en série. Ils remplissent des fonctions clés telles que la catalyse et le métabolisme dans le corps et sont les principaux exécutants des activités de la vie. Les biologistes divisent généralement la structure des protéines en quatre niveaux : la structure primaire décrit la séquence d'acides aminés de la protéine, la structure secondaire se concentre sur la conformation locale de la protéine, la structure tertiaire représente la configuration tridimensionnelle globale de la protéine et la structure quaternaire implique l'interaction entre plusieurs molécules de protéines.Dans le domaine des protéines IA, la recherche repose principalement sur ces structures.
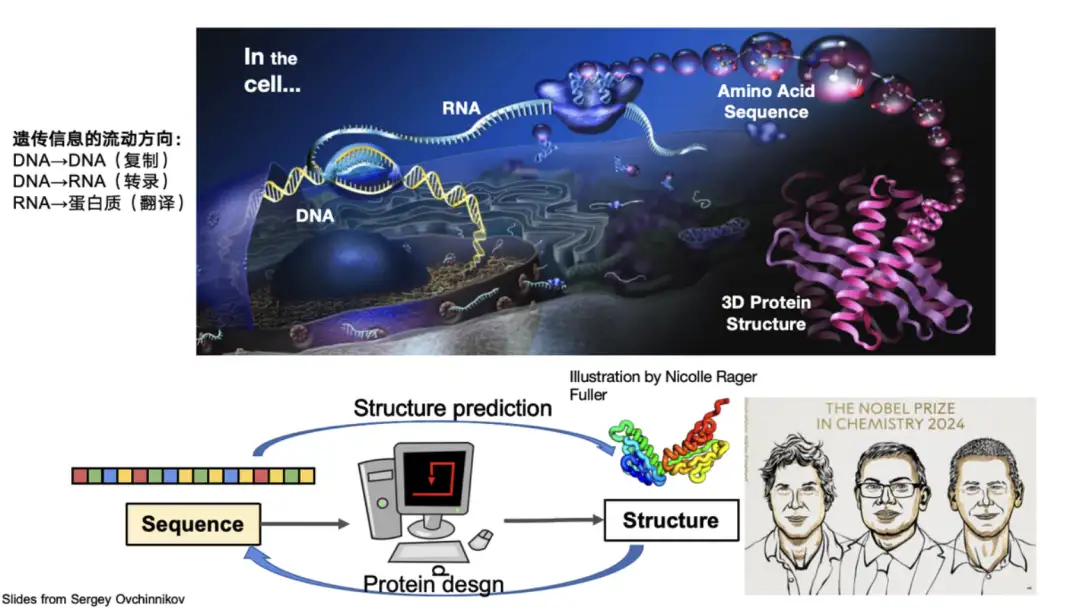
Par exemple, prédire la structure tridimensionnelle d’une protéine à partir de sa séquence est le problème central qu’AlphaFold 2 résout. Il a permis de surmonter le problème du repliement des protéines qui tourmente la communauté scientifique depuis 50 ans et a remporté le prix Nobel pour cela. D'autre part, le professeur David Baker, un contributeur important dans le domaine de la conception des protéines, qui est la conception de nouvelles séquences de protéines basées sur la structure et la fonction, a également remporté le prix Nobel.
Traditionnellement, les structures protéiques sont généralement représentées sous la forme de coordonnées PDB. Ces dernières années, les chercheurs ont exploré des méthodes permettant de convertir des informations de structure spatiale continue en jetons discrets, tels que Foldseek, ProTokens, FoldToken, ProtSSN, ESM-3, etc.
*Foldseek peut coder la structure tridimensionnelle d'une protéine en jetons discrets unidimensionnels.
Le modèle de langage protéique de notre équipe est basé sur ces résultats discrets.
La plupart des recherches sur l'IA et les protéines remontent à la recherche sur le traitement du langage naturel. Examinons donc d'abord deux modèles de langage classiques dans le domaine du traitement du langage naturel (TAL) :L'un est le modèle de langage unidirectionnel représenté par la série GPT,Son mécanisme est basé sur le flux d'informations de gauche à droite, prédisant le prochain jeton en fonction des données de gauche (ci-dessus).L'un est le modèle de langage bidirectionnel représenté par BERT,Il est pré-entraîné via le modèle de langage masqué, qui peut voir les informations (contexte) sur les côtés gauche et droit d'un mot cuit et prédire le mot cuit.
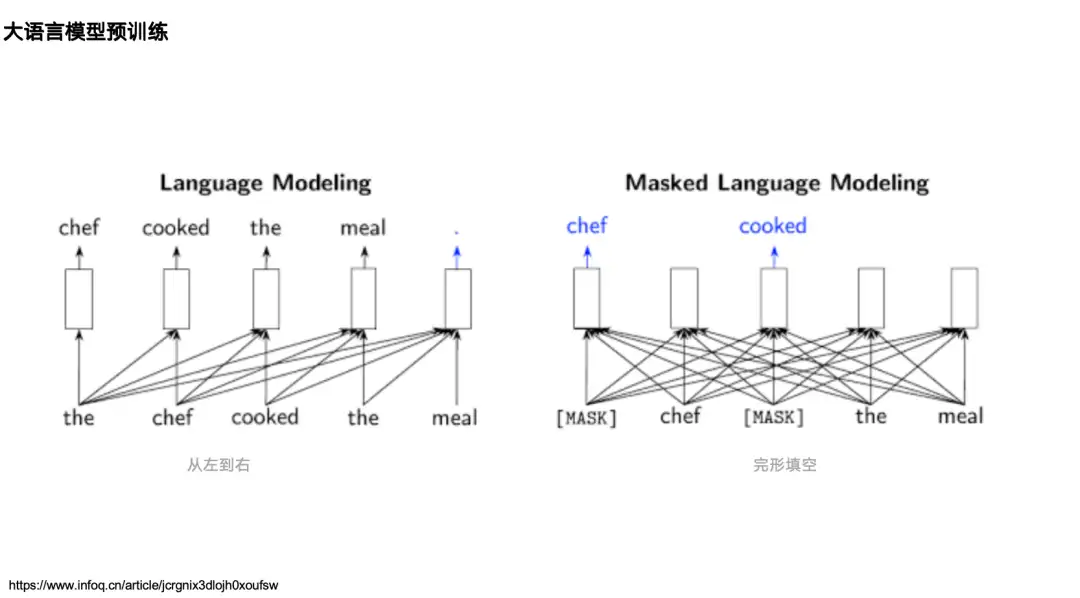
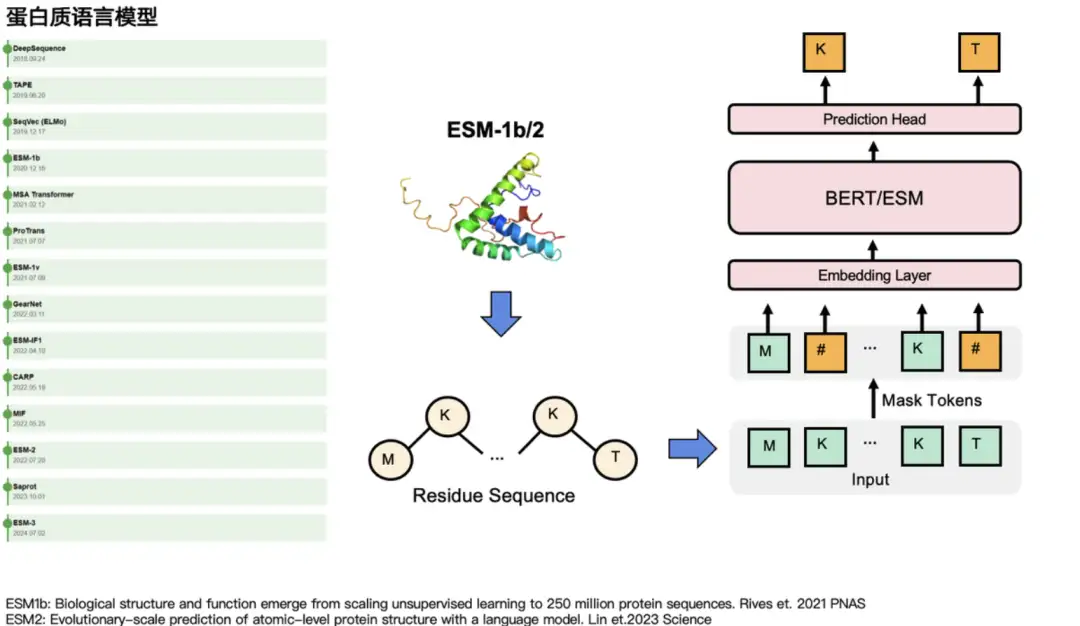
Dans le domaine des protéines, les deux types de modèles ont leurs modèles de langage protéique correspondants.Par exemple, correspondant à GPT, il existe ProtGPT2, ProGen, etc. Correspondant à BERT, il existe les modèles de la série ESM : ESM-1b, ESM-2 et ESM-3. Ils masquent principalement certains acides aminés et prédisent leur « véritable identité ». Dans les tâches de langage naturel, ils masquent certains mots puis les prédisent. Comme le montre le côté gauche de la figure ci-dessous, d'autres modèles de langage qui ont une influence relativement importante dans la communauté des protéines incluent MSA Transformer, GearNet, ProTrans, etc.
Sélectionné pour l'ICLR 2024, le modèle de langage protéique SaProt intègre les connaissances structurelles
Le premier résultat que je souhaite vous présenter est SaProt, un modèle de langage protéique avec un vocabulaire sensible à la structure.Cet article, intitulé « SaProt : modélisation du langage des protéines avec un vocabulaire sensible à la structure », a été sélectionné pour l'ICLR 2024.
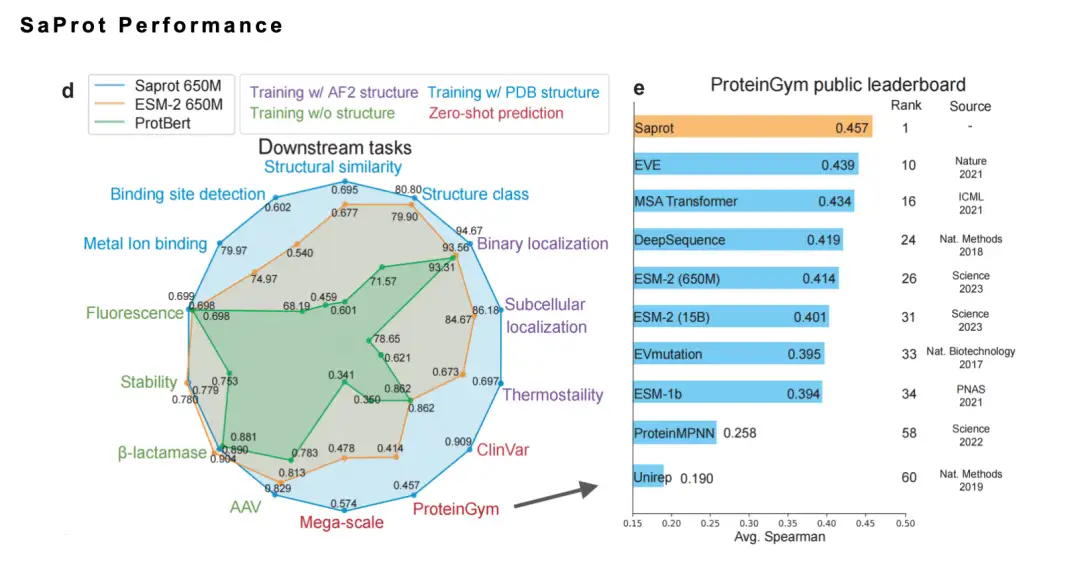
Dans cet article, nous avons proposé le concept de vocabulaire sensible à la structure, combiné des jetons de résidus d'acides aminés avec des jetons de structure et formé un modèle de langage protéique universel à grande échelle SaProt sur un ensemble de données d'environ 40 millions de séquences et de structures protéiques. Ce modèle a largement surpassé les modèles de base matures existants dans 10 tâches en aval importantes.
Adresse open source de SaProt :
https://github.com/westlake-repl/SaProt
Adresse de l'article SaProt :
https://openreview.net/forum?id=6MRm3G4NiU
Pourquoi fabriquons-nous ce modèle ?
En fait, les informations d’entrée de la plupart des modèles de langage protéique sont principalement basées sur des séquences d’acides aminés. Après la percée d'AlphaFold, l'équipe DeepMind a collaboré avec l'Institut européen de bioinformatique (EMBL-EBI) pour publier la base de données de structures protéiques AlphaFold, qui stocke 200 millions de structures protéiques. Nous avons donc commencé à réfléchir : pouvons-nous intégrer les informations structurelles des protéines dans le modèle de langage pour améliorer ses performances ?

Notre approche est très simple : nous utilisons Foldseek pour convertir les informations structurelles de la protéine sous forme de coordonnées en jetons discrets, construisant ainsi un vocabulaire d'acides aminés et un vocabulaire structurel, puis nous combinons ces deux vocabulaires pour générer un nouveau vocabulaire, à savoir le vocabulaire sensible à la structure (jeton SA). De cette manière, la séquence d'acides aminés d'origine peut être convertie en une nouvelle séquence d'acides aminés - dans cette séquence, les lettres majuscules représentent les jetons d'acides aminés et les lettres minuscules représentent les jetons de structure. Ensuite, nous pouvons continuer à travailler sur le modèle du langage masqué. Sur cette base, nous avons formé un modèle SaProt de 650 millions de paramètres à l'aide de 64 GPU A100 avec un temps de formation total d'environ 3 mois.
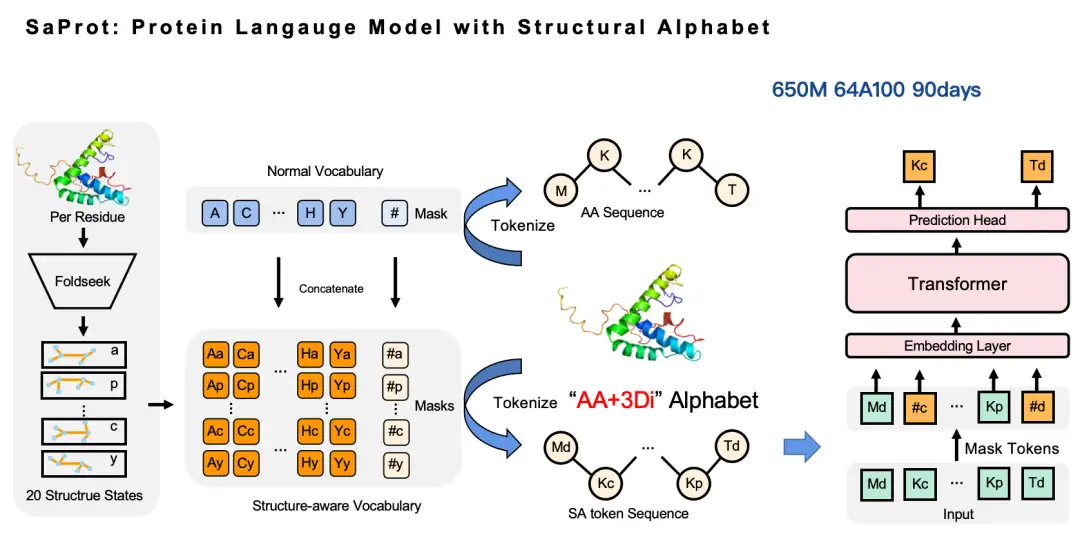
Pourquoi avons-nous choisi Foldseek pour convertir les jetons de structure protéique ?
Il nous a fallu six mois pour finalement décider de la séquence de jetons Foldseek 3Di. Intuitivement, l’intégration d’informations structurelles dans le modèle de langage protéique devrait améliorer les performances, mais lorsque nous l’avons réellement essayé, nous avons essayé différentes méthodes mais avons échoué. Par exemple, nous avons utilisé la méthode GNN pour modéliser la structure des protéines. Étant donné que la structure des protéines est en fait un réseau neuronal graphique, nous souhaitons naturellement modéliser la structure des protéines sous forme de graphique. Nous avons donc adopté la méthode MIF, mais nous avons constaté que le modèle formé a une faible capacité de généralisation et ne peut pas être étendu à la structure réelle du PDB. Après une analyse approfondie, nous pensons que cela peut être dû au fait que la méthode de modélisation utilisant le modèle de langage masqué entraînera des problèmes de fuite d'informations.
En termes simples, la structure protéique prédite par AlphaFold elle-même présente certains biais, modèles et traces de prédiction de l’IA. Lorsque ces données sont utilisées pour former un modèle de langage, le modèle peut facilement capturer ces traces, ce qui permet au modèle de bien fonctionner sur les données de formation, mais d'avoir une faible capacité de généralisation.
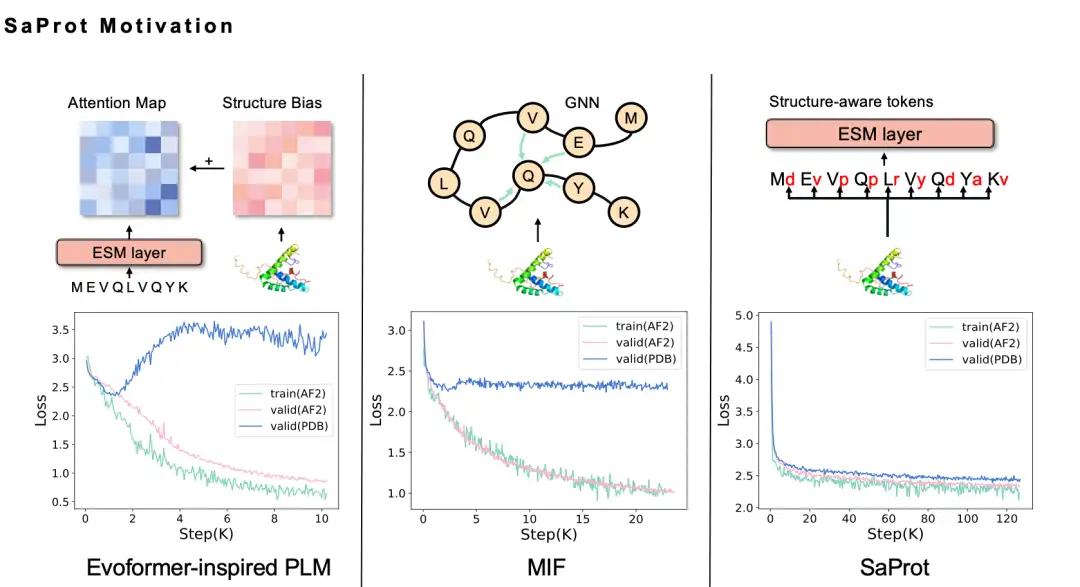
Nous avons essayé diverses améliorations, notamment en utilisant la méthode Evoformer, mais le problème de fuite d’informations existait toujours jusqu’à ce que nous essayions Foldseek. Nous avons constaté que la perte du modèle SaProt obtenue sur les données structurelles prédites par AlphaFold était réduite, et la perte sur les données de structure PDB réelles était également considérablement réduite, ce qui répondait à nos attentes.
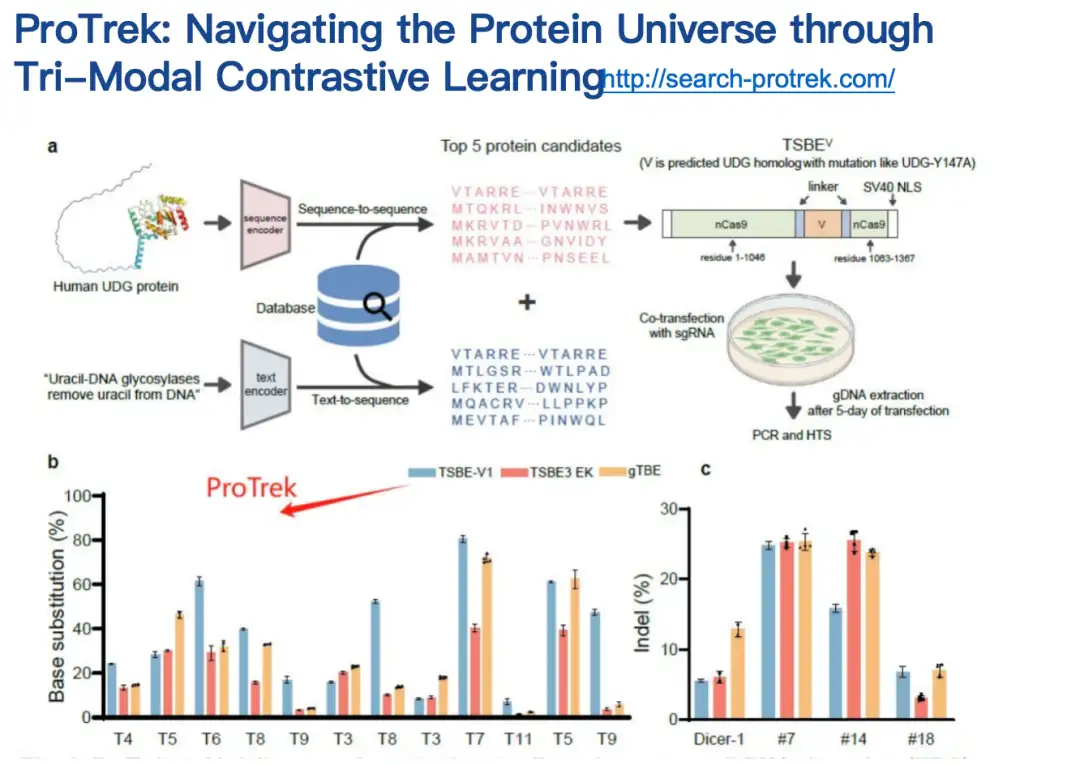
De plus, SaProt obtient de bons résultats sur plusieurs benchmarks.L'année dernière, il s'est également classé premier sur la liste faisant autorité ProteinGym. Dans le même temps, nous avons également collecté les résultats de vérification des expériences humides de la communauté SaProt/ColabSaProt sur plus de 10 protéines (telles que diverses modifications de mutation enzymatique, modification de protéines fluorescentes et prédiction de fluorescence, etc.), qui ont toutes donné d'excellents résultats.
Bien que nous pensons que le modèle SaProt est plutôt bon,Mais étant donné que de nombreux biologistes n’ont pas reçu de formation en apprentissage profond,Il leur est très difficile d’affiner de manière indépendante un modèle de langage protéique comportant environ 1 milliard de paramètres.Nous avons donc construit une plateforme d'interface interactive ColabSaprot + SaprotHub.
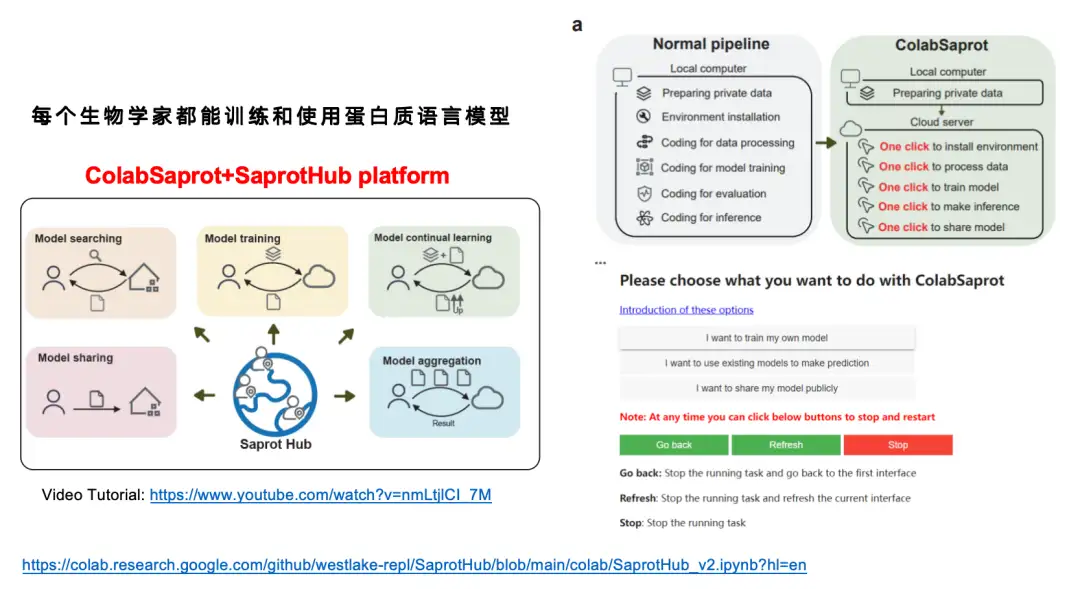
Dans le processus de formation de modèle traditionnel (pipeline normal), les utilisateurs doivent passer par plusieurs étapes, notamment la préparation des données, la configuration de l'environnement, l'écriture de code, le traitement des données, la formation du modèle, l'évaluation du modèle, le raisonnement du modèle, etc. Avec ColabSaprot, l'ensemble du processus a été grandement simplifié - les utilisateurs n'ont qu'à cliquer sur quelques boutons pour terminer l'installation de l'environnement, la formation du modèle, la prédiction et d'autres opérations, réduisant ainsi considérablement le seuil d'utilisation.
Comme le montre la figure ci-dessous, ColabSaprot se compose principalement de trois parties : un module de formation, un module de prédiction et un module de partage.

* Dans le module de formation, les utilisateurs doivent simplement décrire la tâche à gauche et télécharger les données, puis cliquer sur former. Le système sélectionnera automatiquement les hyperparamètres optimaux (tels que la taille du lot, etc.).
* Dans le module de prédiction, les utilisateurs peuvent charger directement leurs modèles précédemment formés et faire des prédictions. Vous pouvez également saisir directement des modèles partagés par d’autres chercheurs pour faire des prédictions.
* Le module de partage fournit un moyen de protéger la confidentialité des données lors du partage des résultats. Les données provenant de nombreux laboratoires sont extrêmement précieuses, et certains chercheurs peuvent avoir besoin d’utiliser ces données pour des recherches de suivi, mais ils souhaitent toujours partager les modèles existants. Dans ColabSaprot, les utilisateurs ne peuvent partager que le modèle lui-même. Étant donné que le modèle est essentiellement une boîte noire, les autres ne peuvent pas obtenir les données originales.
Lors du partage de modèles, étant donné que les modèles de langage sont généralement de grande taille, il est presque impossible de partager directement un modèle avec 1 milliard de paramètres en ligne.Nous avons donc adopté un mécanisme d’adaptateur mature.Les utilisateurs n’ont besoin de partager qu’un très petit nombre de paramètres, généralement seulement 1% ou 1/1 000 des paramètres du modèle d’origine. Tout le monde peut partager des adaptateurs entre eux et charger les adaptateurs d'autres personnes pour effectuer des ajustements précis ou des prédictions en fonction de ceux-ci. Si l’amélioration est bonne, de nouveaux adaptateurs peuvent être à nouveau partagés, formant ainsi un mécanisme de coopération communautaire efficace et améliorant considérablement l’efficacité de la recherche.
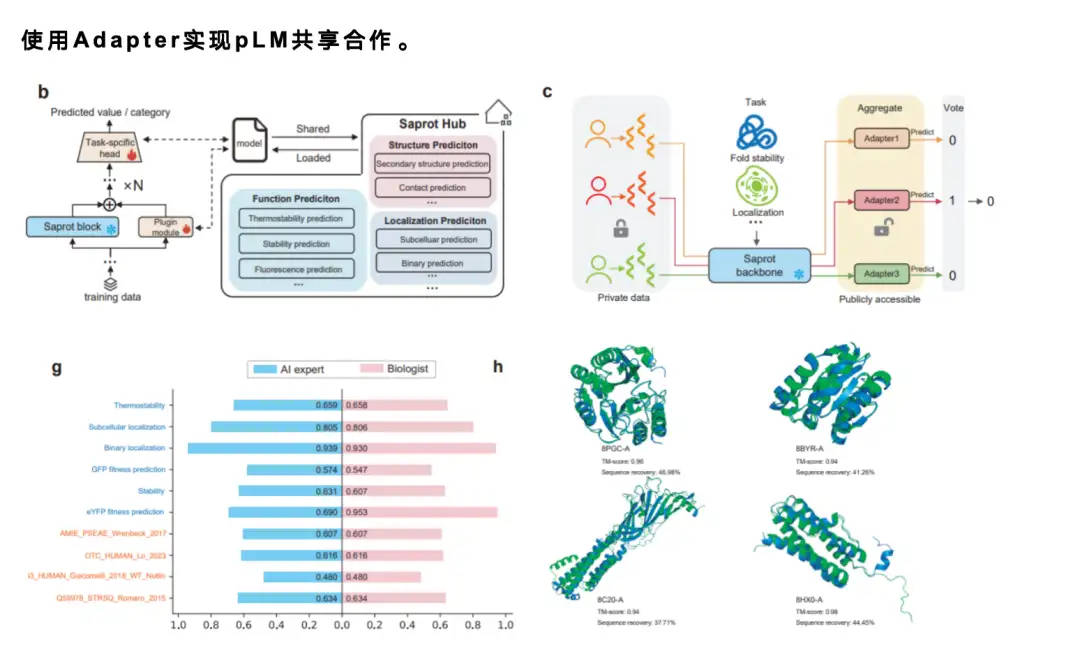
En outre, nous avons également mené une étude auprès des utilisateurs.Nous avons invité 12 étudiants sans expérience en apprentissage automatique ni en programmation à tester la plateforme ColabSaprot. Nous leur avons fourni les données et leur avons indiqué les tâches à effectuer, leur demandant d'utiliser ColabSaprot pour la formation et la prédiction du modèle. Enfin, en comparant leurs résultats avec les performances des experts en IA, nous avons constaté que ces utilisateurs non experts étaient capables d’atteindre un niveau proche de celui des experts utilisant ColabSaprot.
De plus, afin de favoriser le partage de modèles de langage protéique,Nous avons également créé une communauté appelée OPMC,Des chercheurs renommés du pays et de l’étranger dans ce domaine y ont participé, encourageant chacun à partager des modèles et à promouvoir la coopération et la communication.
Adresse de l'OPMC :

Modèle ProTrek : trouver la correspondance entre la séquence, la structure et la fonction des protéines
Le deuxième travail que je souhaite présenter est le modèle de langage protéique ProTrek.
Dans la recherche biologique, de nombreux scientifiques sont confrontés au besoin suivant : ils disposent d’un génome contenant de nombreuses protéines mais ne connaissent pas leurs fonctions spécifiques.
ProTrek est un modèle de langage trimodal pour l'apprentissage contrastif de la séquence, de la structure et de la fonction.Grâce à son interface de recherche en langage naturel, les utilisateurs peuvent explorer le vaste espace protéique en quelques secondes et rechercher des relations entre toutes les combinaisons par paires de séquence, de structure et de fonction pour neuf tâches différentes. En d’autres termes, en utilisant ProTrek, les utilisateurs n’ont qu’à saisir la séquence protéique et cliquer sur un bouton pour trouver rapidement des informations relatives à la fonction et à la structure des protéines. De même, vous pouvez également rechercher des informations de séquence et de structure en fonction de la fonction, et rechercher des informations de séquence et de fonction en fonction de la structure, etc. De plus, il prend également en charge les recherches de classe séquence-séquence et structure-structure.
Adresse d'utilisation de ProTrek :
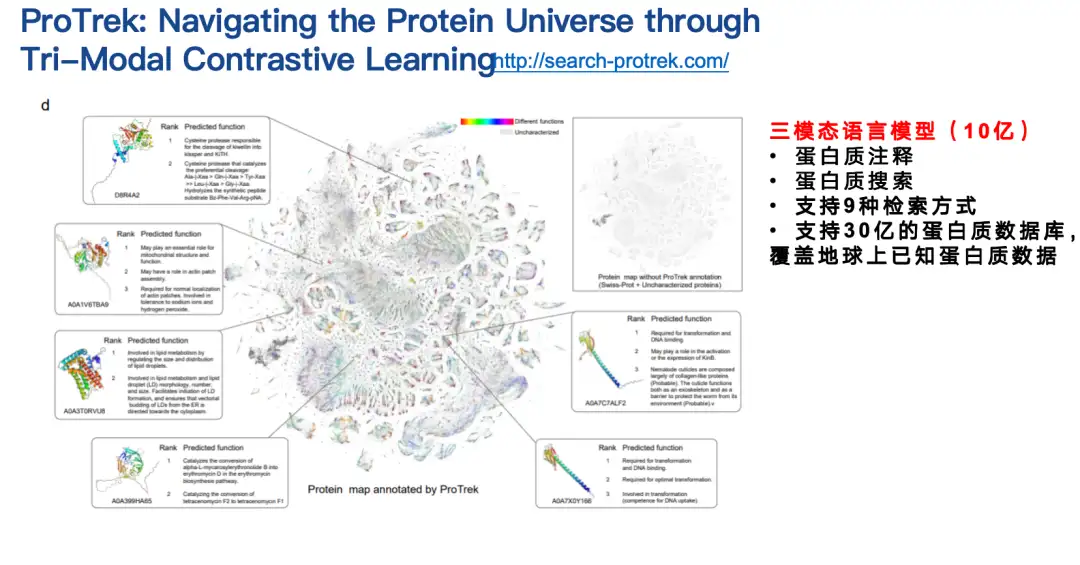
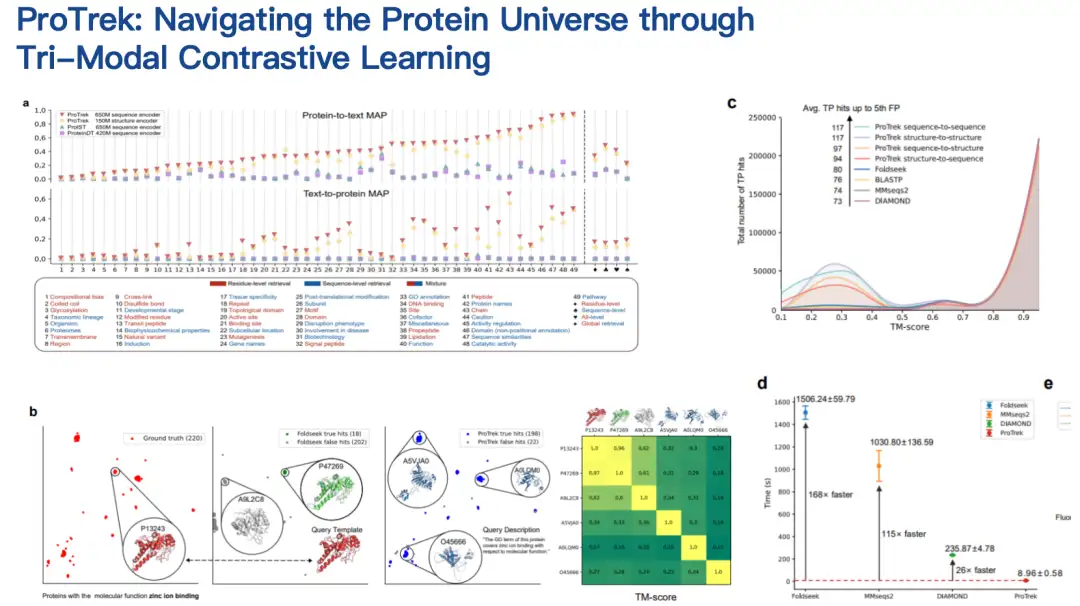
Nos collaborateurs ont évalué le modèle ProTrek dans des tests secs et humides.Par rapport aux méthodes similaires existantes, ProTrek a obtenu des améliorations de performances significatives. De plus, nous avons également utilisé ProTrek pour générer une grande quantité de données pour la formation de notre modèle génératif, qui a également bien fonctionné.
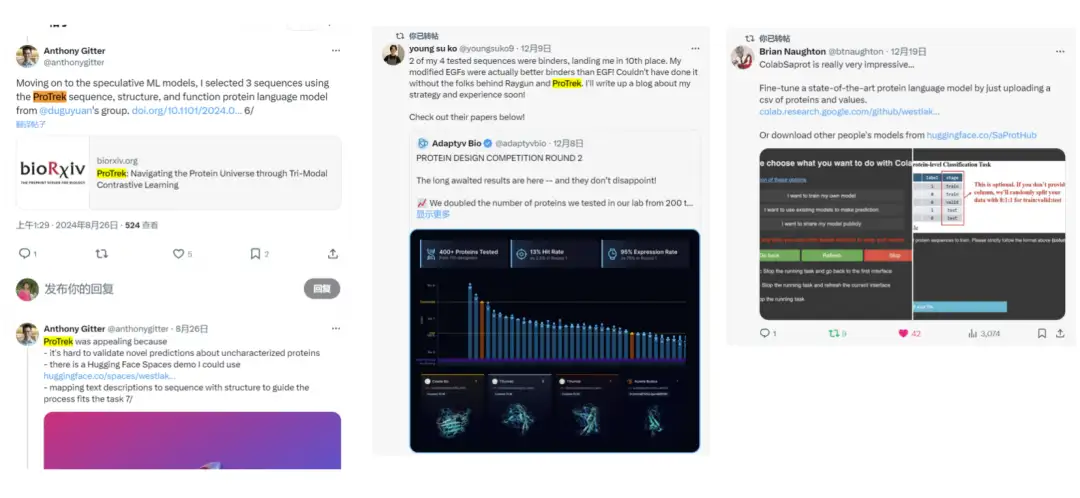
Nous avons remarqué sur Twitter queDe nombreux utilisateurs ont commencé à utiliser ProTrek pour concourir.Nous avons également reçu de nombreux commentaires positifs, ce qui a encore prouvé la praticité du modèle.
Modèle Pinal : concevoir de nouvelles séquences protéiques en saisissant simplement du texte
Un autre de nos travaux est Pinal, un modèle de conception de protéines basé sur des descriptions textuelles.
La conception traditionnelle des protéines doit généralement prendre en compte des facteurs complexes, tels que les informations sur le modèle de fonction énergétique biophysique. Ce que nous voulons explorer, c'est que, puisque les grands modèles de langage fonctionnent bien dans de nombreuses tâches, est-il possible de concevoir un modèle de langage protéique basé sur du texte ? Dans ce modèle, il suffit de décrire simplement les informations d’une protéine pour concevoir sa séquence d’acides aminés ?
Adresse d'utilisation Pinal :
http://www.denovo-pinal.com/
Adresse du document :
https://www.biorxiv.org/content/10.1101/2024.08.01.606258v1
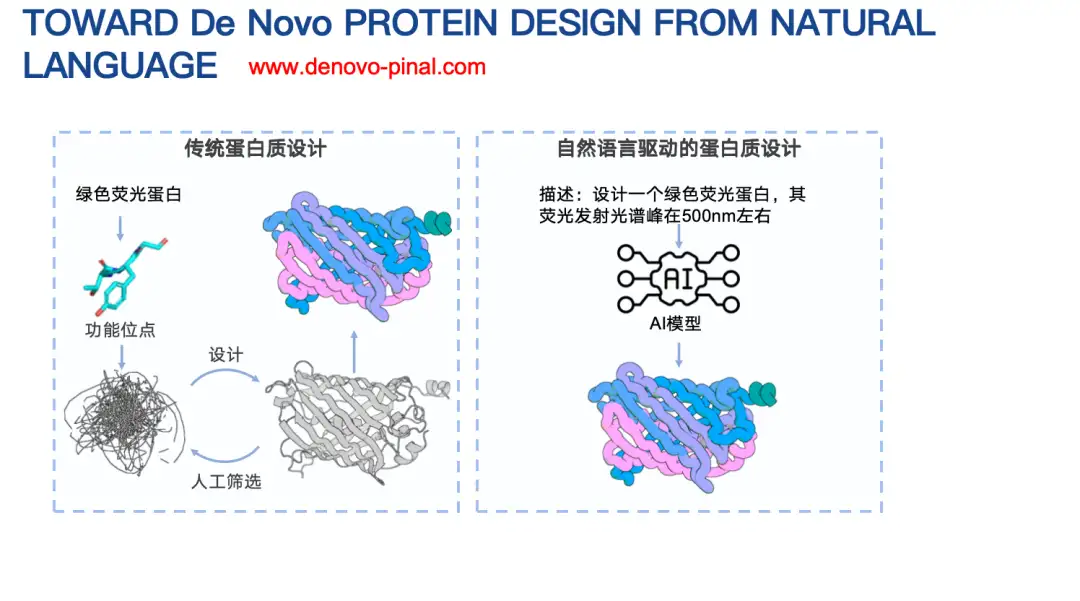
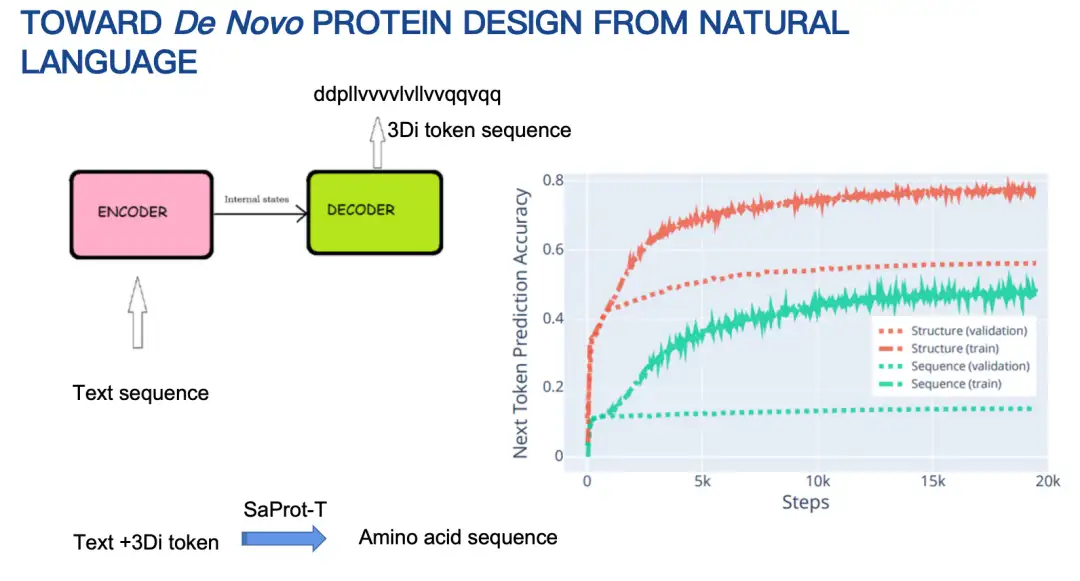
Permettez-moi de présenter brièvement les principes de base de Pinal (16 milliards de paramètres).Au départ, notre idée était d’utiliser une architecture encodeur-décodeur qui entre du texte et produit une séquence d’acides aminés. Cependant, après avoir essayé pendant longtemps, les résultats n’ont pas été idéaux. La raison principale est que l’espace des séquences d’acides aminés est trop grand, ce qui rend les prédictions difficiles.
Nous avons donc ajusté notre stratégie pour concevoir d’abord la structure de la protéine, puis la séquence d’acides aminés en fonction de la structure et des indices textuels. La structure protéique est ici également représentée par un codage discrétisé. Les résultats montrent que la méthode de conception combinée à la structure est nettement plus performante que la méthode de prédiction directe de la séquence d'acides aminés en termes de précision de prédiction du prochain jeton, comme le montre la figure ci-dessous.
Nous avons récemment reçu une vérification en laboratoire humide de Pinal de la part de nos collaborateurs.Pinal a conçu 6 séquences protéiques, dont 3 ont été exprimées et 2 séquences ont été vérifiées comme ayant une activité catalytique enzymatique correspondante. Il convient de mentionner que dans ce travail, nous n’avons pas mis l’accent sur la conception d’une protéine meilleure que le type sauvage. Notre objectif principal est de vérifier si la protéine conçue sur la base du texte possède la fonction protéique correspondante.
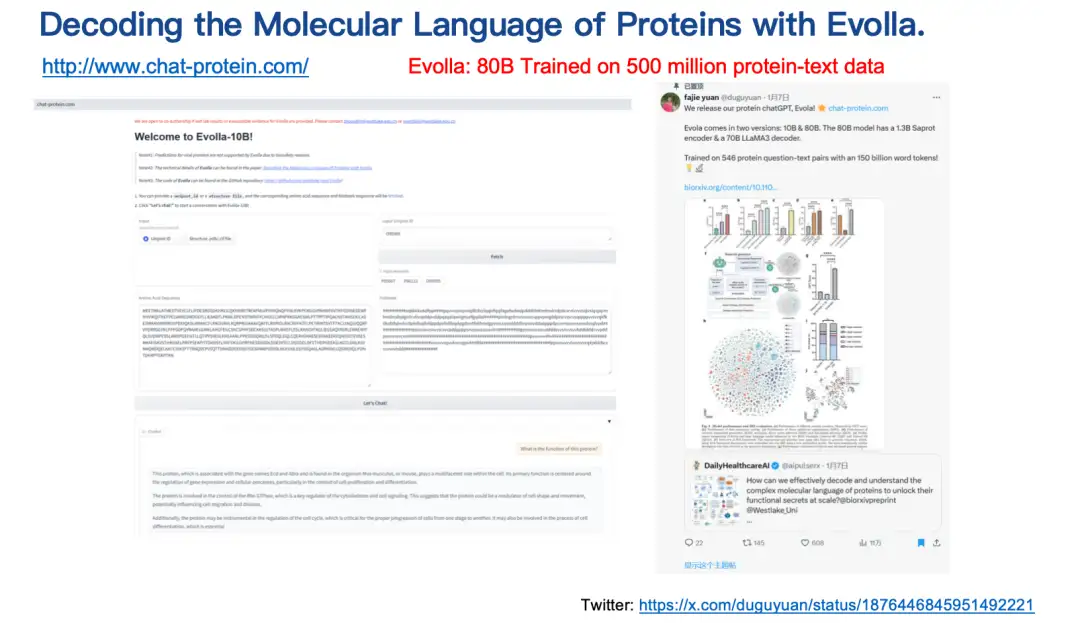
Modèle d'Evolla : Décoder le langage moléculaire des protéines
Le dernier résultat introduit est le modèle Evolla.Il s'agit d'un modèle de génération de langage protéique de 80 milliards de paramètres, l'un des plus grands modèles biologiques open source, conçu pour décoder le langage moléculaire des protéines.
En intégrant la séquence des protéines, la structure et les informations de requête des utilisateurs,Evolla génère des informations précises sur la fonction des protéines.Les utilisateurs n'ont qu'à saisir la séquence et la structure de la protéine, puis à poser des questions, telles que la présentation de la fonction de base ou de l'activité catalytique de la protéine, et à simplement cliquer sur un bouton, et Evolla générera une description détaillée d'environ 200 à 500 mots.
Adresse d'utilisation d'Evolla :
http://www.chat-protein.com/
Adresse du journal Evolla :
https://www.biorxiv.org/content/10.1101/2025.01.05.630192v2

Il convient de mentionner que les données de formation et la puissance de calcul requises pour le projet Evolla sont énormes. Deux de nos doctorants ont passé près d’un an à collecter et à traiter les données de formation. Au final, nous avons généré plus de 500 millions de paires protéines-textes de haute qualité grâce à des données synthétiques, couvrant des centaines de milliards de jetons de mots. Le modèle est assez précis dans la prédiction des fonctions enzymatiques.Mais il y a inévitablement des problèmes d’illusion.
À propos de l'équipe
Le Dr Yuan Fajie de l'Université Westlake est principalement engagé dans la recherche scientifique appliquée liée à l'apprentissage automatique traditionnel et aux sujets interdisciplinaires, et se concentre sur l'exploration des grands modèles d'IA et de la biologie computationnelle. Il a publié plus de 40 articles académiques dans des conférences et revues de premier plan dans le domaine de l'apprentissage automatique et de l'intelligence artificielle (tels que NeurIPS, ICLR, SIGIR, WWW, TPAMI, Molecular Cell, etc.). Pour des informations détaillées sur les membres de l’équipe et les contributeurs du projet, veuillez vous référer au document.
Le groupe de recherche mène des recherches à long terme dans les domaines de l’apprentissage automatique et de l’IA + bioinformatique. Vous êtes invités à postuler pour des postes de doctorants, d'assistants de recherche, de boursiers postdoctoraux et de chercheurs au sein du groupe de recherche. Les étudiants sont invités à visiter le laboratoire pour des stages. Les personnes intéressées peuvent envoyer leur CV à [email protected].








