Command Palette
Search for a command to run...
L'équipe De Zhang Yang À l'Université Nationale De Singapour a Développé Un Algorithme De Prédiction De La Structure De l'ARN De Deuxième Génération Qui a Surpassé SOTA Dans Plusieurs Tests De référence.

La compréhension de la structure et de la fonction des molécules d’ARN a toujours été un axe de recherche essentiel en biologie moléculaire et dans l’industrie pharmaceutique. L'ARN, en particulier l'ARN non codant (ARNnc), peut se replier dans des structures spécifiques et jouer un rôle important dans divers processus cellulaires tels que la régulation des gènes (comme la transcription et la traduction), la catalyse, la transduction du signal biologique et la réponse au stress.
Avec le développement rapide de la technologie de séquençage à haut débit, les données de séquence d’ARN ont augmenté de façon exponentielle, mais l’écart entre les séquences connues et les structures d’ARN résolues expérimentalement se creuse. Il devient donc de plus en plus urgent de résoudre la structure atomique de l’ARN en se basant uniquement sur sa séquence brute. Les chercheurs ont développé diverses méthodes pour étudier la structure de l’ARN, telles que des techniques de biologie structurale telles que la cristallographie aux rayons X, la spectroscopie par résonance magnétique nucléaire et la cryomicroscopie électronique (cryo-EM). Bien que ces techniques expérimentales puissent fournir une résolution plus élevée, l’élucidation expérimentale de la structure tridimensionnelle de l’ARN est souvent coûteuse et, dans certains cas, difficile à réaliser. donc,Il existe une demande croissante de méthodes informatiques permettant de prédire la structure tridimensionnelle de l’ARN de haute qualité directement à partir de la séquence.
La « prédiction de la structure de l'ARN ab initio » fait référence à une méthode qui prédit directement la structure tridimensionnelle de l'ARN à partir de sa séquence sans s'appuyer sur des données expérimentales ou des connaissances préalables. Le cœur de cette méthode consiste à utiliser des techniques de simulation informatique et de chimie computationnelle pour prédire la conformation tridimensionnelle des molécules d’ARN à l’aide de modèles et d’algorithmes mathématiques.
Récemment, les derniers résultats de recherche de l'équipe du professeur Zhang Yang de l'Université nationale de Singapour ont encore promu la « prédiction de la structure de l'ARN ab initio » à un niveau supérieur.Les chercheurs ont proposé un cadre de prédiction de structure d'ARN de haute précision basé sur l'apprentissage profond, DRfold2.Il intègre un modèle de langage composite d'ARN pré-entraîné (RCLM) et un module de structure de débruitage pour la prédiction de la structure de l'ARN de bout en bout. DRfold2 obtient de bons résultats en matière de prédiction de topologie globale et de structure secondaire par rapport à d'autres méthodes de pointe sur plusieurs benchmarks.
Une analyse détaillée montre que cette amélioration provient principalement de la capacité du RCLM à capturer des modèles co-évolutifs et de son processus de débruitage efficace.Cela améliore la précision de prédiction de contact non supervisée de DRfold2 de plus de 100% par rapport aux méthodes existantes.
Les résultats associés ont été publiés sur la plateforme de pré-impression bioRxiv sous le titre « Prédiction de la structure de l'ARN ab initio avec un modèle de langage composite et un apprentissage de bout en bout débruité ».
Points saillants de la recherche :
* DRfold2 intègre un modèle de langage composite d'ARN pré-entraîné (RCLM) et un module de structure de débruitage pour la prédiction de la structure de l'ARN de bout en bout
* Grâce à une combinaison unique de modélisation de langage composite, d'apprentissage de bout en bout basé sur la débruitage et de post-optimisation guidée par l'apprentissage profond, DRfold2 ouvre une nouvelle direction pour la « prédiction de la structure de l'ARN ab initio »
* DRfold2 est hautement complémentaire à AlphaFold3 et permet d'obtenir des améliorations de précision statistiquement significatives après intégration dans le cadre d'optimisation

Adresse du document :
https://www.biorxiv.org/content/10.1101/2025.03.05.641632v1
Télécharger l'ensemble de données de test de structure d'ARN DRfold2 :
Ensemble de données : créer un ensemble de données de test indépendant
Afin d'évaluer objectivement les performances de DRfold2,Les chercheurs ont construit un ensemble de données de test indépendant contenant 28 structures d’ARN.Leurs longueurs de séquence sont toutes inférieures à 400 nt et proviennent des trois catégories suivantes :
* Dernières séquences cibles de RNA-Puzzles
* Séquences cibles d'ARN dans la compétition CASP15
* Les structures d'ARN les plus récemment publiées dans la base de données Protein Data Bank (PDB) au 1er août 2024
Il est à noter que les chercheurs ont exclu les grandes structures d’ARN synthétiques de l’ensemble de données CASP15 car elles s’écartent des structures d’ARN trouvées dans la nature, qui sont au centre de l’analyse fonctionnelle et de la conception de médicaments.
Afin de garantir une évaluation rigoureuse du modèle, l'ensemble de formation ne contient que des structures d'ARN publiées avant 2024 et exclut les ARN dont la similarité de séquence est supérieure à 80% avec l'ensemble de données de test.
Télécharger l'ensemble de données de test de structure d'ARN DRfold2 :
Architecture du modèle : un nouveau pipeline de prédiction de la structure 3D de l'ARN DRfold2
DRfold2 est un nouveau pipeline de prédiction de structure 3D de l'ARN qui se compose de quatre modules principaux : (1) Modèle de langage composite de l'ARN (RCLM), (2) Bloc de transformation de l'ARN, (3) Module de structure de débruitage et (4) Sélection et optimisation du modèle final via le protocole CSOR, comme illustré dans la figure A ci-dessous :
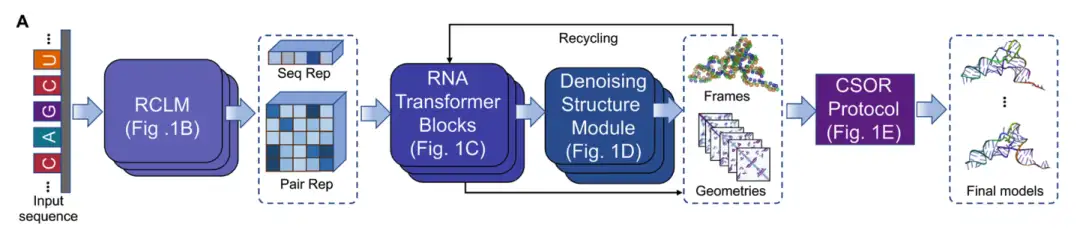
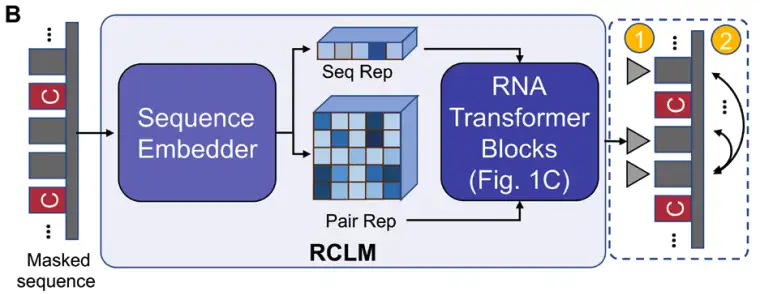
En commençant par une séquence d'ARN d'entrée,DRfold2 code d'abord la séquence de requête à l'aide d'un modèle de langage composite ARN pré-entraîné (RCLM).Générer une représentation de séquence (Seq Rep) et une représentation de paire (Pair Rep) ; Le RCLM est formé sur des données de séquence non supervisées à grande échelle via la méthode de maximisation de la vraisemblance composite pour obtenir une reconnaissance de modèle de séquence plus efficace, comme illustré dans la figure B ci-dessous :
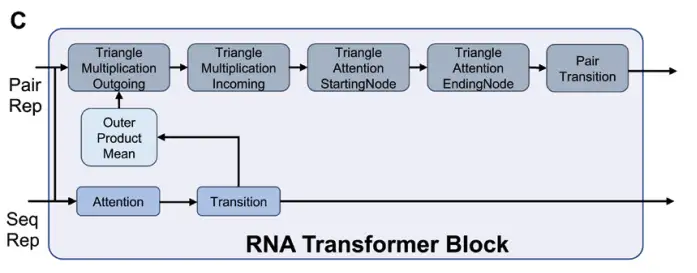
Ces séquences et représentations appariées sont ensuite entrées dans le module RNA Transformer pour être traitées afin de générer des représentations de caractéristiques clés requises pour le repliement de la structure de l'ARN, comme illustré dans la figure C ci-dessous :
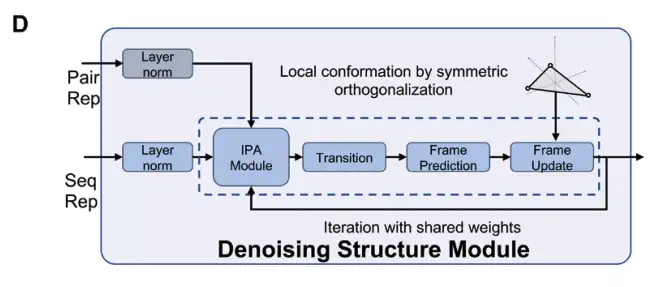
Ensuite, DRfold2 utilise le module de structure d'ARN débruité (DRSM) pour générer des conformations d'ARN de bout en bout, comme illustré dans la figure D ci-dessous :
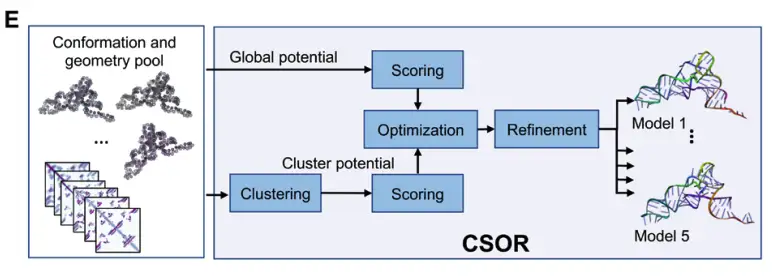
Le modèle final de structure de l'ARN est examiné et optimisé via le protocole CSOR de post-traitement pour sélectionner et affiner le meilleur modèle parmi l'ensemble des conformations générées à plusieurs points de contrôle, comme illustré dans la figure E ci-dessous :
Bien que DRfold2 porte un nom similaire à la méthode DRfold précédente de l'équipe, il introduit des avancées significatives basées sur un cadre complètement différent.L’élément le plus important est l’intégration d’un modèle de langage composite, qui améliore considérablement la capacité de représentation des séquences et des paires d’ARN.De plus, le pipeline de prédiction intègre un module de débruitage de la structure de l'ARN (DRSM), qui utilise une stratégie de perturbation contrôlée pour apprendre de manière robuste les transformations structurelles en corrigeant efficacement les conformations bruyantes de l'ARN.
Les chercheurs ont rendu le serveur en ligne DRfold2 et le code local accessibles au public à l'adresse suivante :
https://zhanglab.comp.nus.edu.sg/DRfold2
Résultats de recherche : DRfold2 surpasse les autres méthodes de pointe sur plusieurs benchmarks
Les chercheurs ont d'abord comparé DRfold2 avec cinq méthodes de prédiction de structure d'ARN de pointe, notamment RNAComposer (basé sur l'assemblage et l'optimisation de fragments), trRosettaRNA (méthode d'apprentissage en profondeur), RhoFold (méthode d'apprentissage en profondeur de bout en bout), RoseTTAFoldNA (méthode d'apprentissage en profondeur de bout en bout) et DeepFoldRNA (méthode d'apprentissage en profondeur).
Comme le montre la figure ci-dessous, les chercheurs ont comparé les résultats de l’évaluation du score TM et du RMSD de DRfold2 et de la méthode de référence à différents seuils de similarité de séquence (50%-80%). Parmi eux, le TM-score est une fonction de notation indépendante de la longueur utilisée pour évaluer la qualité globale de la structure d'ARN prédite. La plage de valeurs est de 0 à 1. Plus la valeur est élevée, plus la similitude entre la structure prédite et la structure réelle est élevée.
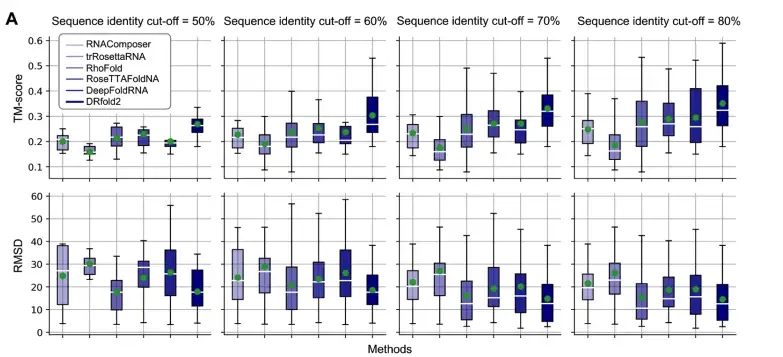
Les résultats montrent que DRfold2 obtient toujours le score TM moyen le plus élevé sous tous les seuils de similarité de séquence.Par exemple:
* Sous le seuil de similarité 80%, le score TM moyen de DRfold2 est de 0,351, soit 18,6% de plus que le deuxième DeepFoldRNA (score TM = 0,296).
* Sous le seuil de similarité 50% (l'ensemble de tests le plus strict), DRfold2 peut toujours obtenir un score TM moyen de 0,269, ce qui est 17,5% supérieur au deuxième RoseTTAFoldNA (score TM = 0,229).
* De plus, le RMSD (écart quadratique moyen) de DRfold2 à tous les seuils de similarité de séquence est toujours inférieur à celui de toutes les méthodes de contrôle, indiquant que sa structure prédite est plus proche de la structure réelle de l'ARN.
Les chercheurs ont également utilisé le ribozyme de type HDV CPEB3 du chimpanzé (PDB ID : 7QR3) comme exemple. L'ARN est long de 69 nucléotides et les effets de prédiction de différentes méthodes sur sa structure tertiaire d'ARN ont été analysés. Les résultats sont les suivants :
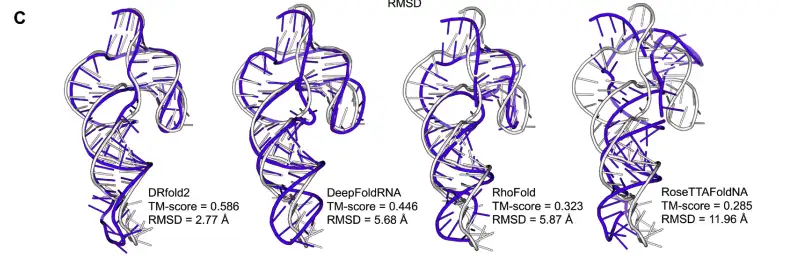
* DRfold2 a capturé avec précision la structure topologique globale du ribozyme, avec un score TM de 0,586 et un RMSD de seulement 2,77 Å.
* DeepFoldRNA fonctionne bien en termes de disposition hélicoïdale globale, mais la direction de la boucle en épingle à cheveux s'écarte considérablement, ce qui entraîne un RMSD aussi élevé que 5,68 Å, soit le double de l'écart de DRfold2.
* RhoFold et RoseTTAFoldNA présentent des erreurs plus importantes dans la prédiction spatiale dans les régions de jonction, ce qui fait chuter le score TM à 0,323 et 0,285.
* La similarité de séquence la plus élevée entre l'ARN cible et l'ensemble de données d'entraînement n'est que de 60,9%, ce qui indique que DRfold2 peut toujours fournir des prédictions de structure fiables pour les nouvelles séquences d'ARN en l'absence de modèles homologues.
Ces résultats montrent que :La représentation probabiliste complète fournie par les modèles de langage d’ordre supérieur comme RCLM améliore considérablement la capacité à apprendre des modèles co-évolutifs et des contraintes spatiales.Ainsi, une modélisation plus précise de la structure de l'ARN 3D a été obtenue grâce au réseau de bout en bout de DRfold2.
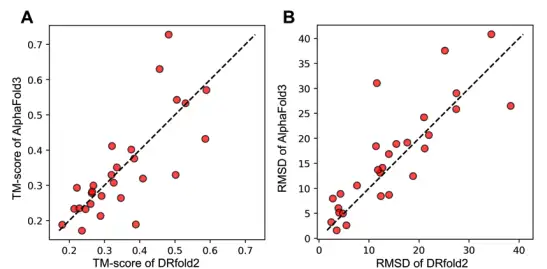
Sur cette base, afin de comparer les performances de DRfold2 et AlphaFold3 dans la prédiction de la structure 3D de l'ARN, les chercheurs ont également soumis les séquences d'ARN de l'ensemble de tests au serveur AlphaFold et ont obtenu la structure prédite d'AlphaFold3 en utilisant la configuration de départ par défaut. s'avérer,Le score TM moyen (0,351) et le RMSD (14,6 Å) de DRfold2 sont légèrement supérieurs à ceux d'AlphaFold3 (0,345 et 16,0 Å).
Ce qui est plus intéressant à noter, c’est que même si DRfold2 et AlphaFold3 présentent des performances globales similaires, les résultats de la figure ci-dessous mettent en évidence la forte complémentarité entre les deux, en particulier lorsque la prédiction s’écarte considérablement de la ligne diagonale.En incorporant les prédictions d’AlphaFold3 comme terme de fonction potentiel supplémentaire dans le cadre d’optimisation DRfold2, les chercheurs ont obtenu des améliorations statistiquement significatives du score TM et du RMSD.
L'équipe du professeur Zhang Yang se concentre sur la recherche en IA et en biologie computationnelle depuis de nombreuses années
Le DRfold2 proposé dans cette étude est en fait une version améliorée du modèle DRfold précédemment proposé par l'équipe du professeur Zhang Yang.
En septembre 2023, l'équipe du professeur Zhang Yang a publié un article intitulé « Intégration de l'apprentissage de bout en bout avec des potentiels géométriques profonds pour la prédiction de la structure de l'ARN ab initio » dans la revue Nature Communications.
Cette étude présente une nouvelle technologie, DRfold, permettant de prédire avec précision la structure tridimensionnelle de l’ARN.L'innovation principale réside dans l'introduction de deux fonctions d'énergie potentielle complémentaires : le potentiel FAPE et le potentiel géométrique.Ils sont formés via deux réseaux de transformateurs indépendants et constituent ensemble un potentiel d'apprentissage profond pour la prédiction de la structure de l'ARN. Les résultats informatiques montrent que par rapport aux méthodes informatiques précédentes de prédiction de la structure de l'ARN, DRfold surpasse ces méthodes dans de nombreux indicateurs de performance.

Adresse du document :
https://www.nature.com/articles/s41467-023-41303-9
De DRfold à DRfold2, l'équipe du professeur Zhang Yang continue de se concentrer sur la recherche en intelligence artificielle et en biologie computationnelle depuis de nombreuses années. Son laboratoire est l’un des premiers à mener des recherches sur la prédiction de la structure des protéines et de l’ARN basées sur l’apprentissage automatique profond. Il a remporté des distinctions telles que le prix Sloan, le prix de carrière de la National Science Foundation et le prix de recherche en sciences fondamentales de l'Université du Michigan. Depuis 2015, il a été sélectionné sept fois dans la liste mondiale des scientifiques les plus cités de Thomson Reuters/Clarivate Analytics. L'algorithme I-TASSER développé par son laboratoire (https://zhanggroup.org/I-TASSER/), Depuis 2006, elle a été classée comme la méthode automatisée de prédiction de la structure des protéines la plus précise dans les expériences mondiales CASP pendant neuf fois consécutives.
Le 2 janvier 2024, l'équipe du professeur Zhang Yang a publié un article intitulé « Améliorer la prédiction des monomères et des structures complexes des protéines d'apprentissage en profondeur à l'aide de DeepMSA2 avec d'énormes données métagénomiques » dans la revue Nature Methods.
L’étude a développé deux nouveaux logiciels pour améliorer la précision de la prédiction structurelle des interactions protéiques. Les auteurs ont développé DeepMSA2, qui utilise une programmation dynamique récursive et des algorithmes de modèle de Markov cachés pour extraire rapidement des données MSA de haute qualité à partir de bibliothèques de séquences métagénomiques massives, puis utilise le logiciel DMFold nouvellement développé pour construire la structure tridimensionnelle du complexe protéique.
Les résultats expérimentaux montrent que la précision de prédiction structurelle de DMFold/DeepMSA2 pour les complexes protéiques est nettement meilleure que celle d'algorithmes tels qu'AlphaFold2. En particulier, DMFold (https://zhanggroup.org/DMFold) a remporté le championnat de prédiction de la structure des complexes protéiques lors du dernier concours de prédiction de la structure des protéines (CASP15).

Adresse du document :
https://www.nature.com/articles/s41592-023-02130-4
Récemment, l’équipe a élargi son orientation de recherche pour inclure la conception et la prédiction de la structure de l’ARN et des peptides courts, et a exploré des sujets liés à la conception de médicaments. À l’avenir, je crois que le professeur Zhang Yang continuera à diriger son équipe pour explorer les mystères de la biologie.
Références :
1.https://www.biorxiv.org/content/10.1101/2025.03.05.641632v1
2.https://mp.weixin.qq.com/s/X_VJ-WOWEP08p5GAJOgq9A








