Command Palette
Search for a command to run...
AlphaFold Franchit Une Nouvelle Étape Dans Son Application ! Une Équipe De l'Université De Cambridge Propose AlphaFold-Metainference Pour Prédire Avec Précision Les Ensembles De Structures Protéiques Désordonnées
Depuis l’émergence d’AlphaFold fin 2018, le domaine de la prédiction de la structure des protéines a connu d’énormes changements avec le soutien de l’IA. Aujourd'hui, AlphaFold impressionne non seulement par sa précision de prédiction, mais élargit également progressivement sa plage de prédiction dans la dernière itération. Il n'est pas étonnant que Shi Yigong, académicien de l'Académie chinoise des sciences, ait un jour généreusement commenté ce projet devant les médias : « À mon avis, c'est la plus grande contribution de l'intelligence artificielle à la science. C'est aussi l'une des avancées scientifiques les plus importantes de l'humanité au XXIe siècle. C'est une réalisation historique remarquable dans l'exploration scientifique du monde naturel par l'humanité. »
Bien que la révolution de la prédiction de la structure des protéines menée par AlphaFold soit si rapide, certains problèmes non résolus se dressent encore sur son chemin. Parmi eux, la recherche sur les protéines désordonnées a toujours été un problème difficile dans le domaine des sciences de la vie. Ces protéines jouent un rôle clé dans la signalisation cellulaire, les processus de régulation et diverses maladies.Cependant, en raison de leur hétérogénéité particulière et de leur dynamique structurelle, ils ne peuvent pas être représentés par une structure unique.Par conséquent, ses recherches n’ont pas fait de progrès aussi significatifs que la prédiction de structures protéiques ordonnées. Le succès d’AlphaFold a ouvert de nouvelles voies pour résoudre les problèmes des scientifiques.
Récemment, une équipe de recherche de l’Université de Cambridge a publié une nouvelle étude et proposé une méthode appelée AlphaFold-Metainference.Cette méthode utilise la corrélation entre la carte d'erreur alignée prédite (PAE) prédite par AlphaFold et la matrice de changement de distance dans la simulation de dynamique moléculaire (MD) pour construire des ensembles structurels de protéines désordonnées et de protéines contenant des régions désordonnées.Il fournit de nouvelles idées pour la prédiction de structures protéiques désordonnées basées sur des méthodes d'apprentissage en profondeur et élargit également le champ d'application d'AlphaFold.
Actuellement, les résultats de recherche pertinents ont été publiés dans la revue académique internationale Nature Communications sous le titre « Prédiction AlphaFold d'ensembles structuraux de protéines désordonnées ».
Points saillants de la recherche :
* Dépassez les limites de la prédiction et obtenez une prédiction de haute précision. L’étude a confirmé qu’AlphaFold peut prédire avec précision les distances entre les résidus même sans être formé sur des données protéiques désordonnées.
* Innover dans les méthodes de prédiction et construire des collections structurées. Cette méthode utilise la distance prédite par AlphaFold comme contrainte structurelle, combinant le cadre de méta-inférence et la simulation de dynamique moléculaire pour construire une collection de structures de protéines désordonnées et de protéines contenant des régions désordonnées.
* Approfondir les méthodes d’apprentissage en profondeur et élargir les limites des applications. Cette méthode fonctionne bien dans le traitement des protéines hautement désordonnées et partiellement désordonnées. L'ensemble de structures généré est significativement plus cohérent avec les données expérimentales qu'une seule structure AlphaFold, résolvant efficacement le problème de la prédiction de la structure des protéines désordonnées.

Adresse du document :
https://www.nature.com/articles/s41467-025-56572-9
Le projet open source « awesome-ai4s » rassemble plus de 200 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : Vérification rigoureuse des données multi-sources
En termes de formation de modèles d'apprentissage en profondeur, étant donné que les collections structurelles de protéines désordonnées sont très faibles en nombre et en précision, mais que les protéines désordonnées peuvent être prédites sur la base des informations disponibles sur les protéines ordonnées, les chercheurs ont utilisé un grand nombre de structures protéiques pliées à haute résolution dans la Protein Data Bank (PDB) pour former des modèles d'apprentissage en profondeur.
En termes de comparaison de données expérimentales, il est difficile d’obtenir des informations expérimentales sur les distances entre les résidus dans les protéines désordonnées, et les étiquettes de données elles-mêmes peuvent affecter les propriétés de l’ensemble conformationnel.Pour ce faire, les chercheurs ont utilisé des données de diffusion des rayons X aux petits angles (SAXS) et des mesures de diffusion par résonance magnétique nucléaire (RMN).Il fournit des informations sans étiquette sur la distribution de distance entre les résidus de protéines désordonnés pour la recherche, qui sont utilisées pour comparer et vérifier les résultats de prédiction.
En outre, pour une vérification plus approfondie,Les chercheurs ont également analysé les données d'ensemble structurelles de l'Aβ et de l'α-synucléine obtenues par des simulations de dynamique moléculaire tous atomes et des simulations à gros grains à l'aide de CALVADOS-2 (C2).Cela vérifie davantage la précision de la distance prédite par AlphaFold.
Architecture du modèle : méthode innovante de méta-raisonnement par fusion
La méthode AlphaFold-Metainference décrite dans cette étude est utilisée pour générer une collection de structures représentant les états natifs de protéines désordonnées et de protéines contenant des régions désordonnées.
Le cœur de l'approche repose sur l'observation que les distances inter-résidus prédites par AlphaFold sont relativement précises même pour les protéines désordonnées et peuvent donc être utilisées comme contraintes structurelles dans les simulations de dynamique moléculaire dans un cadre de méta-inférence. En termes simples, pour générer l'ensemble de structures, AlphaFold-Metainference utilise les distances prédites comme contraintes structurelles dans la simulation de dynamique moléculaire.Convertissez les cartes de distance AlphaFold (distogrammes) en ensembles de structures.
Le premier est la distance de prédiction AlphaFold. Les chercheurs ont utilisé la carte de distance d'AlphaFold pour prédire la distance moyenne entre les résidus et ont calculé la distance prédite et l'écart type à l'aide d'une formule spécifique. Ensuite, un alignement de séquences multiples a été réalisé sur la base de MMseqs2, et la prédiction a été réalisée à l'aide du modèle AlphaFold 1.1.1 avec les paramètres par défaut, sans utiliser de modèle structurel. Les distances inter-résidus générées par AlphaFold sont réparties dans 64 bacs de largeur égale, allant de 2,15625 à 21,84375 Å, le dernier bac comprenant également des distances dépassant 21,84375 Å.
Ensuite, la méthode du méta-raisonnement est combinée. Le méta-raisonnement est une méthode de raisonnement bayésien qui permet de déterminer l'ensemble de structures en combinant des informations préalables et des données expérimentales basées sur le principe d'entropie maximale. À ce stade,Les chercheurs ont utilisé le graphique de distance prédit par AlphaFold comme données pseudo-expérimentales et ont appliqué la méthode de méta-inférence bayésienne.Déterminer l’ensemble structurel en séparant l’hétérogénéité structurelle des erreurs systématiques, telles que les inexactitudes dans le champ de force ou le modèle direct, les erreurs aléatoires dans les données et les erreurs dues à la taille limitée de l’échantillon dans l’ensemble.
Dans les simulations de dynamique moléculaire, les calculs sont effectués sur la base de la fonction d'énergie de méta-inférence et les paramètres d'erreur sont déterminés par le biais de simulations de répliques multiples et d'échantillonnage de Gibbs.Enfin, le champ de force CALVADOS-2 a été utilisé pour réaliser une simulation à gros grains.Implémenter AlphaFold-Metainference.
La dernière étape est la sélection des contraintes de distance. À cette étape, la distance prédite par AlphaFold est filtrée en fonction de la probabilité de distance et de l’erreur d’alignement prédite.Les critères de sélection ont été déterminés en combinant l'hydrophilie des protéines et les scores du test de différence de distance locale prédite (pLDDT).Il convient de noter que l’utilisation expérimentale des scores pLDDT pour sélectionner les distances résiduelles dans les régions structurées n’exclut pas leur utilisation comme contraintes de distance pour optimiser la génération d’ensembles de structures.
Toutes les simulations de dynamique moléculaire ont commencé à partir de la structure prédite par AlphaFold et ont été réalisées sous l'ensemble NVT. Six répliques ont été configurées pour chaque simulation, chaque réplique a fonctionné pendant 1 million d'étapes et la simulation a démarré à partir de différentes positions initiales obtenues lors de l'étape de minimisation de l'énergie.La simulation utilise un intégrateur de Langevin.Le pas de temps est de 5 fs, le coefficient de frottement est de 0,01 ps⁻¹ et un modèle basé sur Cα avec des paramètres CALVADOS-2 et une forme fonctionnelle est utilisé.
Parmi eux, pour les protéines hautement désordonnées et partiellement désordonnées, PULCHRA a été utilisé pour convertir toutes les structures de la collection à gros grains en représentations entièrement atomiques, puis GROMACS a été utilisé pour la minimisation de l'énergie afin d'obtenir des structures plus précises.
Dans l’ensemble, les résultats présentés par les chercheurs illustrent comment les méthodes d’apprentissage profond développées à l’origine pour prédire l’état natif des protéines repliées peuvent être utilisées pour générer un ensemble de structures qui représentent l’état natif des protéines désordonnées. Cette méthode élargit considérablement la portée de la prédiction de la structure des protéines basée sur l’apprentissage profond et fournit une nouvelle idée pour la prédiction de la structure désordonnée des protéines.
Résultats expérimentaux : vérifier pleinement sa rationalité
En termes de précision de prédiction AlphaFold
Les chercheurs ont comparé un ensemble de 11 protéines pour lesquelles des mesures de diffusion SAXS et RMN étaient disponibles, et ont trouvé une bonne concordance entre les distributions de distance prédites par AlphaFold et les distributions de distance dérivées de SAXS. Les chercheurs ont également ajouté une protéine repliée comme contrôle, comme le montre la figure ci-dessous.
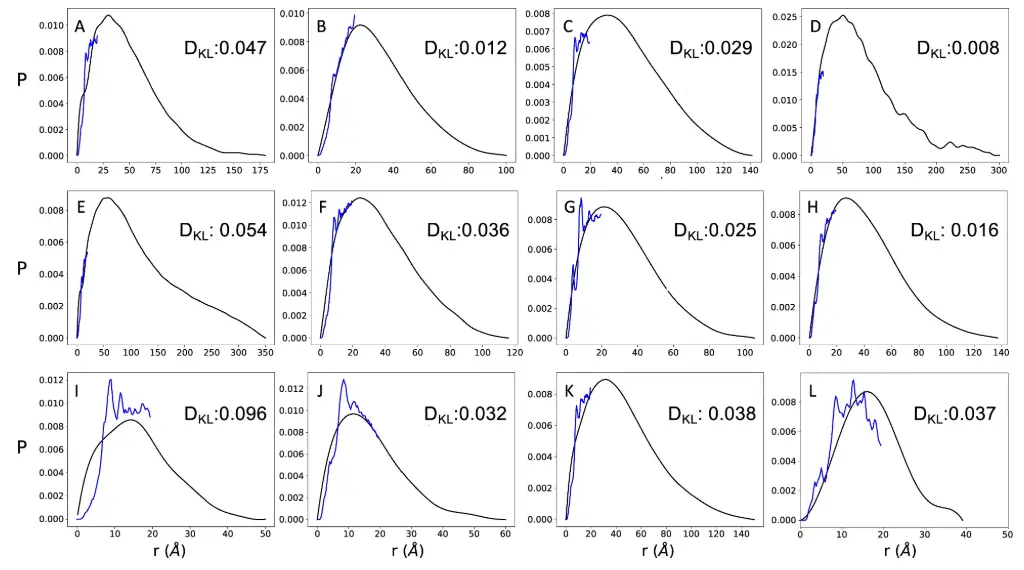
Il convient de mentionner que la distribution de distance prédite par AlphaFold ne couvre pas l'intégralité de la distribution dérivée de SAXA, puisque la distance prédite par AlphaFold peut atteindre environ 22 Å. Les résultats ont montré que la valeur DKL du groupe témoin ajouté était de 0,037, ce qui était comparable aux valeurs DKL de 11 protéines hautement désordonnées (la plage DKL était de 0,008 à 0,096).Cela démontre en outre qu'AlphaFold a une précision comparable dans la prédiction des distances inter-résidus pour les protéines désordonnées et ordonnées.
De plus, les distances prédites par AlphaFold présentent également une bonne concordance avec les distances rétrocalculées à partir des ensembles MD d'Aβ et d'α-synucléine et de l'ensemble CALVADOS-2.
Dans la vérification des collections de structures hautement désordonnées
La distribution de distance par paires peut être calculée à l'aide de mesures de diffusion des rayons X aux petits angles. Les chercheurs ont comparé la distribution de distance obtenue expérimentalement avec la distribution de distance obtenue à partir de l'ensemble des structures déterminées par les simulations AlphaFold-Metainference, à nouveau pour les 11 protéines hautement désordonnées mentionnées ci-dessus.
Dans le même temps, pour une comparaison plus approfondie, les chercheurs ont également montré la distribution de distance obtenue à l'aide de CALVADOS-2, ainsi que la distribution de distance dérivée d'AlphaFold générée directement à partir d'une seule structure AlphaFold. Pour fournir une comparaison quantitative, les chercheurs ont constaté que l'ensemble des structures fournies par AlphaFold-Metainference avec CALVADOS-2 était plus cohérent avec les données SAXS qu'une seule structure dérivée d'AlphaFold.
Les chercheurs ont ensuite comparé les ensembles structurels à l'aide de décalages chimiques RMN, qui ont été rétrocalculés à chaque pas de temps à l'aide de CamShift.Les résultats montrent que dans certains cas, les prédictions d’AlphaFold-Metainference sont plus précises.Comme le montre la figure ci-dessous.
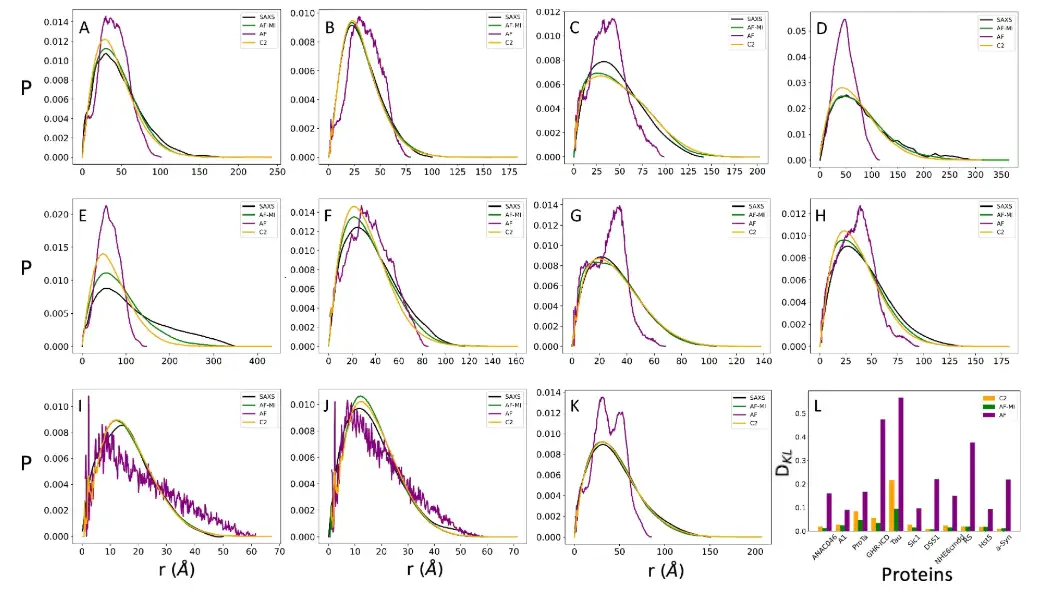
* La distribution des distances expérimentales par paires obtenues par SAXS est représentée par une ligne noire
* La prédiction de structure unique AlphaFold est représentée par une ligne violette
* Les prédictions d'ensemble de structure AlphaFold-Metainference sont représentées par des lignes vertes
* La distribution de distance par paires obtenue par CALVADOS-2 est représentée par une ligne orange
Dans la vérification des collections structurées partiellement désordonnées
Les chercheurs ont préparé un ensemble de six protéines avec des domaines ordonnés et désordonnés, avec des longueurs de séquence différentes et pour lesquelles des données SAXS étaient disponibles pour vérification.
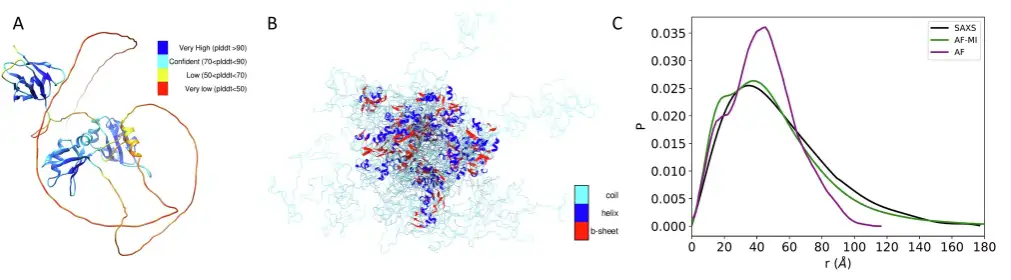
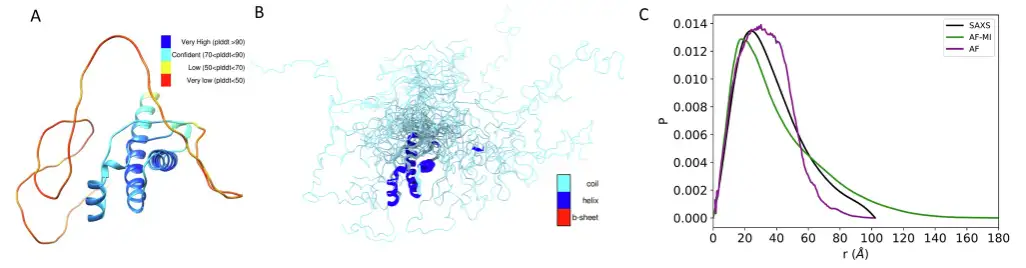
La première est TDP-43, une protéine multifonctionnelle de liaison à l’ARN dotée d’une structure modulaire qui participe à une variété de processus cellulaires, notamment la transcription, l’épissage du pré-ARNm et la régulation de la stabilité de l’ARNm, qui a été impliquée dans la SLA et d’autres maladies neurodégénératives.
Les résultats expérimentaux ont montré qu'en appliquant les critères de filtrage des chercheurs pour sélectionner les distances prédites par AlphaFold, puis en appliquant AlphaFold-Metainference avec ces contraintes de distance,L'ensemble structurel obtenu est en bien meilleure concordance avec les données SAXS.La valeur DKL n'est que de 0,018.C'est mieux que la valeur DKL de 0,582 lors de l'utilisation de la structure prédite AlphaFold directement avec les données SAXS.Comme le montre la figure ci-dessous.
Les chercheurs ont ensuite analysé l’ataxine-3 et la protéine prion humaine. Pour le premier, des résultats similaires ont été obtenus comme pour TDP-43 décrit ci-dessus, où la structure prédite obtenue en utilisant AlphaFold directement à partir de la base de données de structure protéique AlphaFold était peu cohérente avec les données SAXS, avec une valeur DKL de 0,653, alors que lorsqu'un critère de filtrage a été appliqué pour sélectionner les distances prédites par AlphaFold à partir de la simulation AlphaFold-Metainference,Un ensemble de structures plus cohérentes avec les données SAXS a été obtenu.La valeur DKL est seulement de 0,020. Comme le montre la figure ci-dessous.

Pour ces derniers, la structure prédite obtenue directement à partir de la base de données de structures protéiques AlphaFold en utilisant AlphaFold est peu cohérente avec les données SAXS, avec une valeur DKL de 0,1,Lorsque des critères de filtrage ont été appliqués, un ensemble de structures a été obtenu qui était plus cohérent avec les données SAXS.La valeur DKL n'est que de 0,053. Comme le montre la figure ci-dessous.
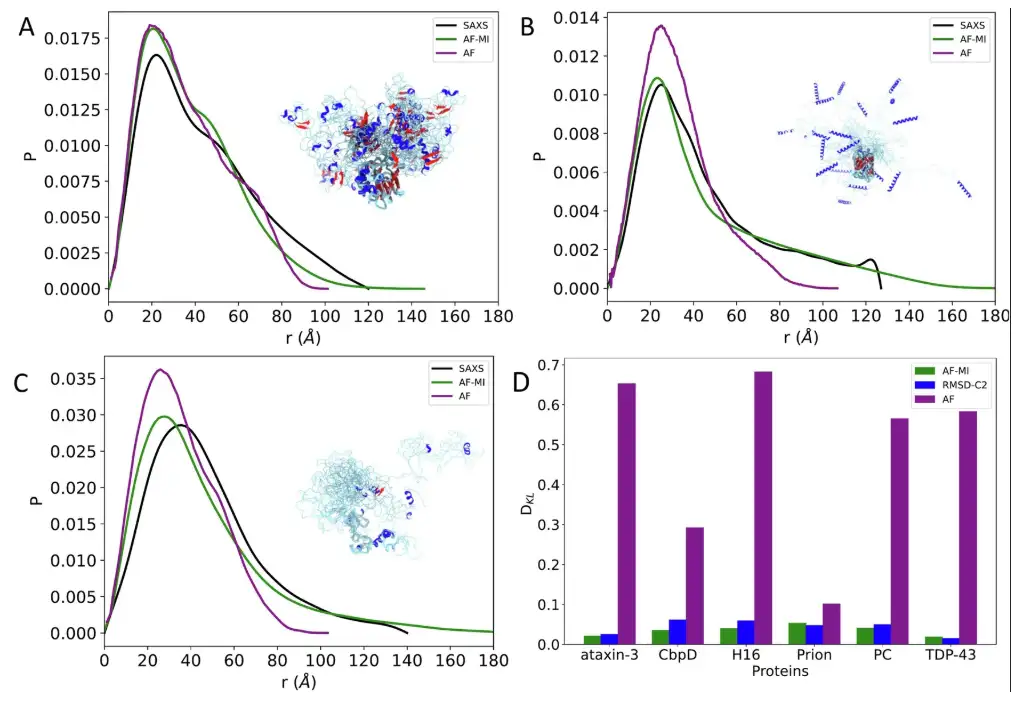
En outre, les chercheurs ont également étudié trois autres protéines, CbpD, H16 et PC. Les résultats ont montré queDans tous les cas, la concordance entre les distributions de distance inter-résidus expérimentales et rétrocalculées est très bonne.Il s’agit d’une amélioration significative par rapport à la structure unique AlphaFold obtenue directement à partir de la base de données de structures protéiques AlphaFold, comme le montre la figure D ci-dessous.
Enfin, en comparaison avec la méthode CALVADOS-2, AlphaFold-Metainference a obtenu de meilleurs résultats sur quatre des six protéines (ataxine-3, CbpD, H16 et PC) et a produit des ensembles structurels comparables sur les deux autres (TDP-43 et protéine prion humaine). Comme le montre la figure ci-dessous.
Progrès dans la prédiction des protéines désordonnées basées sur l'apprentissage profond
Au cours des dernières années, AlphaFold a été principalement utilisé pour prédire la structure statique des protéines repliées, ce qui lui a également valu des critiques de la part de la communauté de recherche scientifique. Cette étude confirme sans aucun doute qu’elle présente également des avantages potentiels en termes d’application dans la prédiction de structures protéiques désordonnées, et fournit également une nouvelle direction de recherche pour la prédiction de structures protéiques désordonnées.
En fait, avec l’intégration étroite de l’IA et des sciences de la vie,De nombreuses discussions ont eu lieu sur la prédiction des structures protéiques désordonnées.L’utilisation de l’IA pour révéler les mystères de la vie est également devenue une méthode courante dans le domaine des sciences de la vie modernes.
Par exemple, un article publié précédemment dans Current Opinion in Structural Biology a discuté des progrès de l’application de l’apprentissage profond dans la recherche sur les protéines intrinsèquement désordonnées (IDP) et a expliqué son rôle dans la promotion de la prédiction des protéines désordonnées et de la caractérisation des ensembles conformationnels.
La recherche connexe a été publiée sous le titre « Apprentissage profond pour les protéines intrinsèquement désordonnées : des prédictions améliorées au déchiffrement des ensembles conformationnels ».
* Adresse du papier :
https://www.sciencedirect.com/science/article/pii/S0959440X24001775
Par coïncidence, une équipe de recherche de l'Université de Copenhague au Danemark a publié un article sur la recherche sur les protéines désordonnées dans Nature intitulé « Ensembles conformationnels du protéome intrinsèquement désordonné humain ». L'article a discuté de l'utilisation de diverses méthodes d'apprentissage en profondeur pour prédire les régions désordonnées, les ensembles conformationnels et les propriétés associées des IDP, y compris les méthodes d'apprentissage en profondeur telles qu'AlphaFold mentionnées ci-dessus, ainsi que les modèles de langage protéique, les réseaux antagonistes génératifs, etc.
*Adresse papier :
https://www.nature.com/articles/s41586-023-07004-5
Il ne fait aucun doute que le développement rapide de l’IA accélère notre compréhension du véritable sens de la vie. Il a fallu 12 ans au scientifique britannique John Kendrew pour utiliser la cristallographie aux rayons X afin d'explorer la première structure protéique. Il ne faudra désormais plus que quelques années à AlphaFold pour percer le mystère du repliement de centaines de millions de protéines. À l’avenir, qui pourra affirmer que nous ne pouvons pas maîtriser la prédiction des structures protéiques désordonnées ?








