Command Palette
Search for a command to run...
De Nouveaux Traitements Contre Le Cancer Pourraient Être Développés ! L'Université Duke Utilise PepPrCLIP Pour Résoudre Le Problème Des Médicaments « Non Médicamenteux »
En 2021, OpenAI a publié le modèle révolutionnaire CLIP (Contrastive Language-Image Pre-training). Grâce à l’apprentissage non supervisé, CLIP peut comprendre et associer efficacement la relation entre les images et le texte sans avoir besoin d’informations d’annotation supplémentaires.
Quelques années plus tard, un groupe de scientifiques biomédicaux s'en est inspiré : puisque CLIP fait correspondre les images et le langage, la même idée peut-elle être utilisée pour faire correspondre les peptides et les protéines ?
En s’appuyant sur les recherches révolutionnaires d’OpenAI sur la génération d’images réalistes grâce à un pré-entraînement contrastif langage-image,Une équipe de recherche du département de génie biomédical de l'université Duke a construit le pipeline PepPrCLIP (Peptide Prioritization by CLIP), qui peut concevoir de courtes protéines (peptides) capables de se lier et de détruire des protéines pathogènes auparavant non médicamenteuses.Comparé à RFDiffusion, une plateforme existante qui génère des peptides à l'aide d'une structure 3D cible, PepPrCLIP est plus rapide et crée des peptides qui correspondent presque toujours plus étroitement à la protéine cible. Les chercheurs ont également vérifié par des expériences que les « peptides guides » sélectionnés par PepPrCLIP peuvent atteindre une liaison et une régulation ciblées robustes et supérieures lorsqu'ils sont utilisés comme peptides inhibiteurs in vitro ou fusionnés avec le domaine de l'ubiquitine ligase E3.
Les résultats correspondants ont été publiés dans Science Advances en janvier de cette année sous le titre « Conception de novo de liants peptidiques pour des cibles conformationnellement diverses avec modélisation linguistique contrastive ».
Adresse du document :
https://www.science.org/doi/10.1126/sciadv.adr8638
Adresse de téléchargement du jeu de données associé :
https://go.hyper.ai/AT5m9
Le projet open source « awesome-ai4s » rassemble plus de 200 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Une nouvelle approche pour résoudre le problème de l'incurabilité des médicaments
Une approche pour traiter la maladie consiste à développer des médicaments capables de cibler et de détruire spécifiquement les protéines responsables de la maladie. Parfois, ces protéines clés ont des structures bien définies, comme des grues en papier soigneusement pliées, de sorte que les thérapies conventionnelles à base de petites molécules peuvent facilement s'y lier.
Cependant, la protéine pathogène de plus de 80% ressemble davantage à un « fouillis emmêlé », désordonné et emmêlé, ce qui rend presque impossible pour les thérapies standard de trouver des sites de liaison à sa surface et de fonctionner. Le terme « non médicamentable » est souvent utilisé pour décrire les protéines dans le développement de médicaments traditionnels qui sont difficiles à devenir des cibles médicamenteuses en raison de leurs caractéristiques structurelles et fonctionnelles.
Selon les informations publiques, les cibles difficiles à droguer présentent souvent les caractéristiques suivantes :
* ont une interface fonctionnelle étendue et plate, dépourvue d’une poche de liaison de ligand bien définie ;
* Absence de ligands spécifiques permettant à la protéine cible de fonctionner ;
* La cible est un inhibiteur de la maladie, nécessitant un médicament pour activer l’activité protéique, ce qui rend le développement de médicaments plus difficile ;
* Les cibles non médicamenteuses ont souvent des fonctions physiologiques complexes, ce qui augmente la difficulté de conception et de développement de médicaments ;
* Limites des stratégies de développement de médicaments.
Pour contourner ces problèmes, de nombreux chercheurs ont étudié comment utiliser des peptides pour se lier aux protéines responsables de maladies et les dégrader. Étant donné que les peptides sont des versions miniatures des protéines, ils ne nécessitent pas de poches de surface pour se lier ; au lieu de cela, les peptides peuvent se lier à différentes séquences d'acides aminés dans les protéines.
Mais même ces approches ont leurs limites, car les liants « prêts à l’emploi » existants ne sont pas conçus pour se fixer à des structures protéiques instables ou trop enchevêtrées. Bien que les scientifiques travaillent dur pour développer de nouvelles protéines de liaison, ces approches reposent toujours sur la cartographie des informations structurelles 3D de la protéine cible, qui ne sont pas disponibles pour les cibles désordonnées.
L’équipe de recherche du département de génie biomédical de l’Université Duke présentée dans cet article a adopté une approche différente. Au lieu d’essayer de cartographier la structure des protéines pathogènes, ils se sont inspirés de grands modèles de langage (LLM) et ont construit PepPrCLIP. Son premier composant, PepPr, utilise un algorithme génératif formé sur une grande bibliothèque de séquences de protéines naturelles pour concevoir de nouvelles protéines « guides » avec des caractéristiques spécifiques ; le deuxième composant, CLIP, utilise un cadre algorithmique développé à l'origine par OpenAI pour tester et déterminer si ces peptides peuvent correspondre à la protéine cible.
Construction d'un processus de priorisation des peptides basé sur CLIP - PepPrCLIP
Comment PepPrCLIP a-t-il été construit ?
En bref, les chercheurs ont d’abord utilisé le modèle de langage protéique ESM-2 (pLM) pour effectuer des perturbations de bruit gaussien sur les inclusions de séquences réelles de liants peptidiques afin de générer des séquences peptidiques candidates avec des caractéristiques naturelles ; ensuite, ces peptides candidats ont été criblés dans l'espace latent via une architecture d'apprentissage contrastif basée sur CLIP pour former un modèle qui co-code des paires peptide-protéine complémentaires ; enfin, le PepPrCLIP construit a intégré un cadre de génération-discrimination pour éliminer des séquences peptidiques candidates complètement nouvelles, capables de se lier à la séquence cible.
La figure suivante montre le processus spécifique de formation du modèle PepPrCLIP :
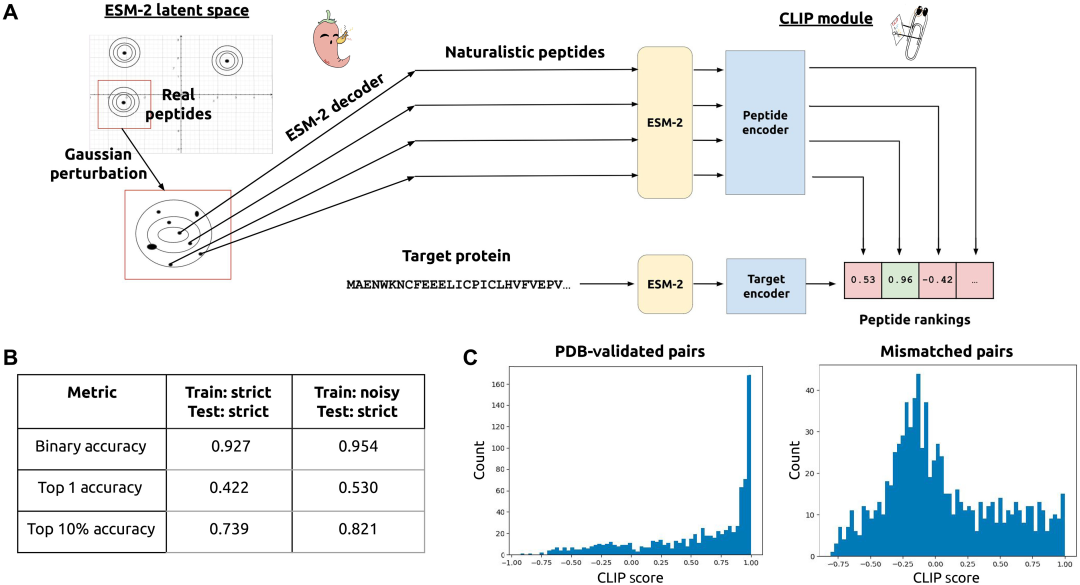
Comme le montre la figure ci-dessus, le peptide naturel intégré dans ESM-2 est échantillonné pour générer une distribution gaussienne, qui est ensuite décodée en une séquence d'acides aminés. Le module CLIP formé code conjointement l'intégration peptide-protéine correspondante, sélectionne des milliers de peptides et évalue leur activité de liaison spécifique à la cible, en particulier :
* Architecture et formation CLIP
Tout d’abord, la séquence d’entrée est intégrée via un modèle ESM-2-650M gelé pour produire une intégration d’entrée ; ensuite, l'intégration d'entrée est moyennée sur la longueur de la séquence pour obtenir un vecteur d'intégration, qui convient aux peptides et aux protéines ; Les couches MLP sont appliquées et le vecteur d'intégration est traité à l'aide de la fonction d'activation de l'unité linéaire rectifiée (ReLU) pour obtenir l'intégration de sortie. Le score CLIP est obtenu en effectuant un produit scalaire entre les incorporations de vecteurs peptidiques et protéiques, avec une valeur comprise entre -1 et 1. Le modèle est formé de manière à ce que les paires de liaison peptide-protéine aient des scores CLIP élevés.
* Génération de séquences peptidiques candidates
Les peptides candidats sont générés à partir de tous les peptides de l'ensemble d'entraînement, chacun d'entre eux étant intégré à l'aide du pLM ESM-2-650M dans PyTorch ; pour un enrobage peptidique donné, la variance de toutes les dimensions de l'enrobage est calculée ; pour chaque résidu dans le peptide source, le bruit est échantillonné à partir d'une distribution normale standard et multiplié par la variance pour créer une perturbation, qui est ajoutée à l'intégration de son résidu correspondant. Au moment de l'inférence, les peptides sources ont été échantillonnés de manière aléatoire à partir de l'ensemble d'entraînement, et pour chaque peptide source, 1 000 peptides ont été générés à l'aide de la méthode de bruit décrite ci-dessus. Enfin, ces peptides (environ 100 000) sont introduits dans le modèle CLIP et classés en fonction de la liaison prévue à la séquence cible fournie par l'utilisateur.
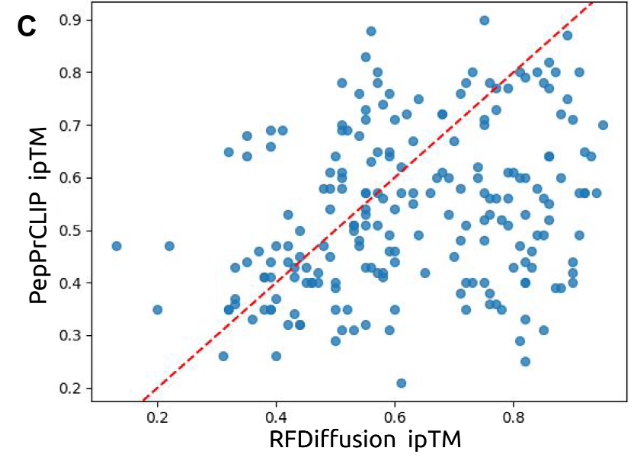
Dans des simulations informatiques, les chercheurs ont comparé les performances de PepPrCLIP avec celles de RFDiffusion. Les chercheurs ont comparé les scores ipTM des peptides générés par PepPrCLIP et ceux générés par RFDiffusion et ont constaté que PepPrCLIP surpassait RFDiffusion pour les peptides avec 33% sur la cible, comme le montre la figure ci-dessous. De plus, en utilisant uniquement l'intégration de séquences, PepPrCLIP est capable d'augmenter considérablement la vitesse de génération et de priorisation, générant environ 1 000 peptides par minute et classant 100 000 peptides par cible protéique en environ 1 minute ; en comparaison, RFDiffusion prend environ 2 minutes pour concevoir un seul liant.Cette efficacité rend PepPrCLIP particulièrement avantageux pour le criblage de grandes bibliothèques de peptides, avec ou sans informations structurelles.
Pour évaluer davantage les effets de PepPrCLIP sur les cibles protéiques ordonnées et désordonnées, l'équipe de recherche a également collaboré avec des équipes de recherche de la Duke University School of Medicine, de l'Université Cornell et du Sanford Burnham Prebys Medical Discovery Institute pour tester expérimentalement la plateforme.
Lors du premier test, l’équipe de recherche a montré queLes peptides générés par PepPrCLIP peuvent se lier efficacement et inhiber l'activité d'UltraID, une protéine enzymatique relativement simple et stable.
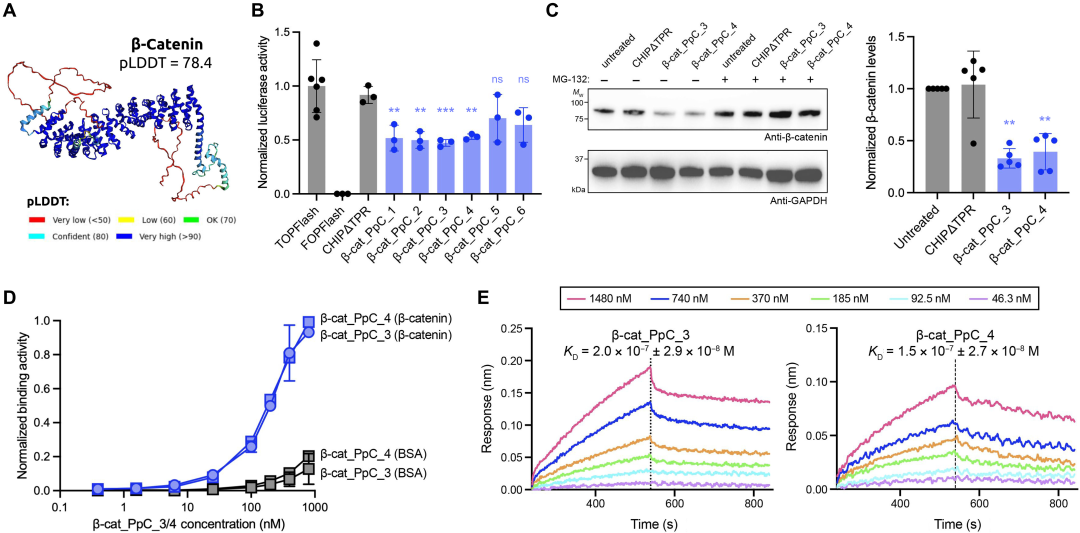
Ensuite, ils ont utilisé PepPrCLIP pour concevoir des peptides capables de se fixer à la β-caténine, une protéine complexe et désordonnée impliquée dans la signalisation dans plusieurs types de cancer différents. Comme le montre la figure ci-dessous, l’équipe a généré six peptides dont CLIP a montré qu’ils pouvaient se lier aux protéines et a montré que quatre d’entre eux pouvaient se lier efficacement à leurs cibles et les dégrader. En perturbant les protéines, ils peuvent ralentir la signalisation des cellules cancéreuses.
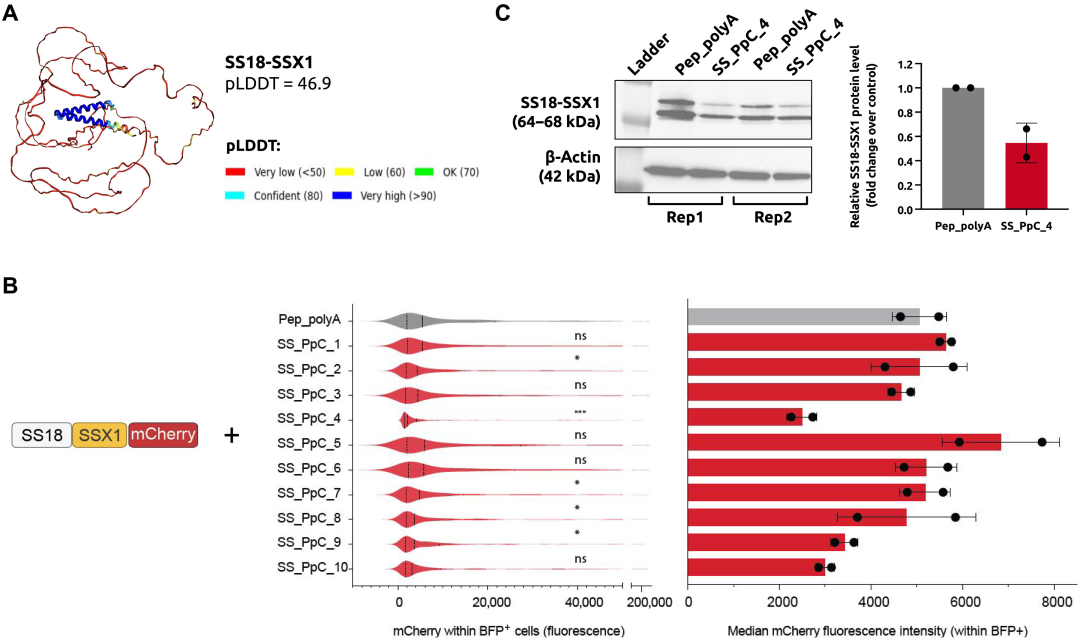
Dans le test le plus complexe, l’équipe a conçu des peptides capables de se lier à des protéines hautement désordonnées associées au sarcome synovial, un cancer malin rare qui représente 5% à 10% de toutes les tumeurs des tissus mous. Le sarcome synovial se développe dans les tissus mous et touche principalement les enfants et les jeunes adultes. La maladie est caractérisée par la présence d’une protéine de fusion oncogène unique et hautement désordonnée, SS18-SSX.
L’équipe a placé les peptides dans des cellules de sarcome synovial et a testé 10 modèles. Comme le montrent les résultats ci-dessous, parmi les peptides prédits par PepPrCLIP pour se lier à SS18-SSX1, SS_PpC_4 a significativement réduit la fluorescence de SS18-SSX1-mCherry ; ensuite, les chercheurs ont également testé l'effet de la surexpression de SS_PpC_4 sur le niveau de protéine de fusion SS18-SSX1 endogène. Il convient de noter que la surexpression du peptide SS_PpC_4 a considérablement réduit les niveaux de protéines SS18-SSX1 (> 40%).
Autrement dit,PepPrCLIP conçoit des peptides capables à la fois de se lier aux protéines et de les dégrader.S’ils parviennent à détruire la protéine, les chercheurs auront la possibilité de développer une thérapie contre des cancers jusqu’alors impossibles à traiter, ce qui ouvre de nombreuses possibilités cliniques passionnantes.
L'IA fournit de nouveaux outils pour vaincre les maladies « incurables »
La dernière revue publiée dans une revue Nature en septembre 2023 présente de manière exhaustive les derniers progrès en matière de découverte de médicaments ciblant les protéines « non médicamenteuses » et leur application clinique.Différentes molécules présentant des caractéristiques similaires non médicamenteuses ont été divisées dans les catégories suivantes :
1 Petites GTPases : telles que les protéines de la famille RAS, notamment KRAS, HRAS et NRAS, qui sont considérées comme non médicamenteuses en raison de l'absence de poches ciblables sur leurs surfaces ;
2 Phosphatase : Étant donné que chaque phosphatase présente de nombreuses similitudes de structure, elle présente une faible sélectivité et des effets secondaires inévitables, ce qui entrave considérablement les progrès de la découverte de médicaments.
③ Facteurs de transcription (TF) : Diverses maladies humaines sont associées à une dérégulation des TF impliqués dans de nombreux processus biologiques, dont la plupart ne peuvent pas être ciblés par les petites molécules conventionnelles en raison de leur hétérogénéité structurelle et de l'absence de sites de liaison traitables.
④ Cibles épigénétiques : Les cibles épigénétiques jouent un rôle essentiel dans la régulation des modèles d’expression génétique et ont un impact sur divers processus biologiques et maladies ;
⑤ Autres protéines : Les interactions protéine-protéine (IPP) et leurs réseaux sont d’une grande importance dans les processus biologiques et la régulation du cycle cellulaire. Certains IPP dotés de surfaces d'interaction plates sont plus difficiles à cibler que d'autres IPP, ce qui les rend « non médicamenteux » dans une certaine mesure.
Aujourd’hui, face aux cibles dites « inmédicamentables », la communauté académique a développé des dizaines de méthodes innovantes. Selon le mécanisme des protéines non médicamenteuses, ils ont adopté des technologies de pointe telles que la découverte de médicaments basée sur des fragments (FBDD), la conception de médicaments assistée par ordinateur (CADD), le criblage virtuel (VS), les bibliothèques codées par l'ADN (DEL), etc. pour former une stratégie systématique de conception de médicaments. Aujourd’hui, le développement de la technologie de l’intelligence artificielle et l’essor des modèles de langage à grande échelle basés sur les protéines ont fourni de nouveaux outils pour surmonter ce problème.Ces dernières années, des avancées importantes ont été réalisées tant dans l’industrie que dans le monde universitaire.
Industrie,En décembre 2023, Absci Corporation, leader dans la découverte d'anticorps par IA générative, a annoncé une collaboration avec AstraZeneca pour développer des anticorps conçus par IA contre une cible tumorale. Cette collaboration combine la plateforme de création de médicaments intégrée d'Absci avec l'expertise d'AstraZeneca en oncologie pour accélérer la découverte de nouveaux candidats potentiels au traitement du cancer. La plateforme de création de médicaments intégrés d'Absci génère des données propriétaires en mesurant des millions d'interactions protéine-protéine, qui sont utilisées pour former les modèles d'IA propriétaires d'Absci et, dans les itérations ultérieures, valider les anticorps conçus à l'aide de nouveaux modèles d'IA. La plateforme accélère la découverte de médicaments en réalisant la collecte de données, la conception pilotée par l'IA et la validation en laboratoire en environ 6 semaines, et a le potentiel d'élargir la gamme de cibles médicamenteuses, y compris le développement de médicaments pour des cibles auparavant considérées comme non médicamenteuses.
milieu universitaire,En janvier 2025, une étude menée conjointement par le leader pharmaceutique de l'IA Insilico Medicine et l'Université de Toronto au Canada a combiné des modèles informatiques quantiques avec des modèles informatiques classiques et de l'intelligence artificielle générative pour explorer un plus large éventail de possibilités chimiques grâce à la formation, la génération et le criblage d'énormes ensembles de données, et a découvert de nouvelles molécules ciblant la protéine motrice du cancer KRAS « non médicamenteuse ».
La mutation KRAS est l’une des mutations les plus courantes dans le cancer, survenant dans environ un quart des tumeurs humaines. La mutation KRAS peut conduire à une prolifération cellulaire incontrôlée et donc au cancer. Dans cette étude, afin de générer de nouveaux inhibiteurs potentiels de KRAS, les chercheurs ont proposé un modèle de cadre hybride quantique-classique qui combine un modèle génératif variationnel quantique (QCBM) et un réseau de mémoire à long terme (LSTM), combinant l'informatique quantique avec des méthodes informatiques classiques pour concevoir de nouvelles molécules. Cette recherche a également été soutenue par plusieurs institutions de recherche, dont le St. Jude Children's Research Hospital. Les résultats de recherche pertinents ont été publiés dans Nature Biotechnology sous le titre « Un algorithme amélioré par l'informatique quantique dévoile des inhibiteurs potentiels de KRAS ».

Grâce aux avancées technologiques, l’humanité a ouvert la voie à un nouvel espace d’imagination et à des possibilités illimitées pour vaincre les maladies.








