Command Palette
Search for a command to run...
Publié Dans La Revue Nature ! Une Équipe De l'Université De Pékin Utilise l'IA Pour Prédire La Direction De l'évolution Des Virus COVID-19/SIDA/grippe, Avec Une Amélioration De La Précision De 67%
En décembre 2019, la pandémie de COVID-19 a soudainement éclaté. Cette maladie causée par le virus SARS-CoV-2 est très contagieuse. En seulement un mois, le nombre de cas dans mon pays a dépassé les 1 000 et s’est rapidement propagé dans le monde entier.
Afin de lutter contre la propagation du virus, notre pays a lancé début 2021 une politique de vaccination universelle gratuite. Cependant, même avec le soutien des vaccins, la crise sanitaire est devenue de plus en plus compliquée.C’est parce que le virus SARS-CoV-2 continue de muter.S'adaptant progressivement à la pression immunitaire générée par le vaccin et aux conditions environnementales changeantes, la « souche virale » découverte à l'origine à Wuhan a disparu depuis longtemps et a été remplacée par diverses souches mutantes, qui continuent de déclencher une nouvelle vague d'infection, et son impact se poursuivra jusqu'après 2023.
Par coïncidence, tout récemment, le taux de positivité du virus de la grippe a continué d’augmenter et de nombreuses personnes ont été infectées par la grippe A (A19) sans le savoir. Semblable au virus SARS-CoV-2, le virus A19 est également très contagieux, se propage rapidement et mute rapidement. Plusieurs sous-types du virus peuvent apparaître au cours de la même saison, ce qui augmente également le risque d’infection répétée dans la population sur une courte période.
Cela montre que prédire la direction de l’évolution du virus est crucial pour la prévention et le contrôle ainsi que pour la conception de vaccins et de médicaments.Cependant, la mutation, en tant que base de l'évolution du virus, est hautement aléatoire, de sorte qu'en général, seul un très petit nombre de mutations peut « simplement » augmenter l'adaptabilité du virus. Ce déséquilibre entre les échantillons positifs (mutations bénéfiques) et les échantillons négatifs (mutations nocives) rend extrêmement difficile la formation d’un modèle d’apprentissage profond capable de prédire les mutations bénéfiques rares du virus. Dans le même temps, les virus ne mutent souvent que sur quelques sites, ce qui rend difficile pour les réseaux neuronaux de capturer directement les faibles changements dans les interactions intramoléculaires causés par les mutations, et pose également des problèmes de modélisation.
À cet égard, le professeur Tian Yonghong et le professeur associé Chen Jie de l'École d'ingénierie de l'information de l'Université de Pékin, en collaboration avec le chercheur Zhou Peng du Laboratoire national de Guangzhou, ont guidé l'étudiant au doctorat Nie Zhiwei et l'étudiant en master Liu Xudong pour réexaminer le problème de la prédiction de l'évolution virale et ont proposé un cadre de prédiction de la force motrice des mutations virales axées sur l'évolution E2VD.Ce cadre peut prédire la direction évolutive du virus SARS-CoV-2, du virus de la grippe, du virus Zika et du VIH (virus du SIDA), améliorant considérablement la vitesse de réponse humaine aux infections virales émergentes et fournissant un soutien important pour l'optimisation rapide des vaccins et des médicaments.
La recherche a été publiée dans Nature Machine Intelligence le 17 janvier 2025 sous le titre « Un cadre d'apprentissage profond unifié axé sur l'évolution pour la prédiction des facteurs de variation des virus ».

Adresse du document :
https://www.nature.com/articles/s42256-024-00966-9
Adresse du document : Suivez le compte officiel et répondez « Viral Evolution » pour obtenir le PDF complet
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : ensemble de données de pré-formation UniRef90 et ensemble de données d'analyse des mutations virales en profondeur
Les virus génèrent continuellement de nouvelles mutations et les accumulent de manière sélective au cours du processus d’évolution. Par conséquent, le modèle de langage protéique pour les scénarios évolutifs doit avoir de fortes capacités de généralisation à échantillon zéro, c'est-à-dire qu'il doit être capable de gérer des mutations invisibles. Pour y parvenir,L'équipe de recherche a choisi UniRef90 comme ensemble de données pour la pré-formation du modèle de langage protéique. UniRef90 contient des informations évolutives riches au niveau de la séquence sans affecter négativement les performances dans les premières étapes de la formation du modèle. Ces riches informations évolutives permettent au modèle d'être exposé à suffisamment d'échantillons de séquences de familles de protéines pendant la pré-formation, améliorant ainsi sa capacité de généralisation à échantillon zéro.
De plus, pour soutenir le modèle d'apprentissage du paysage de fitness évolutif causé par les mutations virales,L’équipe de recherche a utilisé des ensembles de données open source d’analyse des mutations profondes de divers virus.
Architecture modèle : conception architecturale universelle inspirée de l'évolution
Sur la base des modèles d'« amplification de mutation faible » et d'« extraction de mutations bénéfiques rares », l'équipe de recherche a proposé le cadre de prédiction de la force motrice des mutations virales axées sur l'évolution E2VD. Comme le montre la figure a ci-dessous,Il comprend principalement 3 modules :Il s'agit du codage de séquences protéiques, du couplage de dépendance locale-globale et de l'apprentissage focal multitâche.
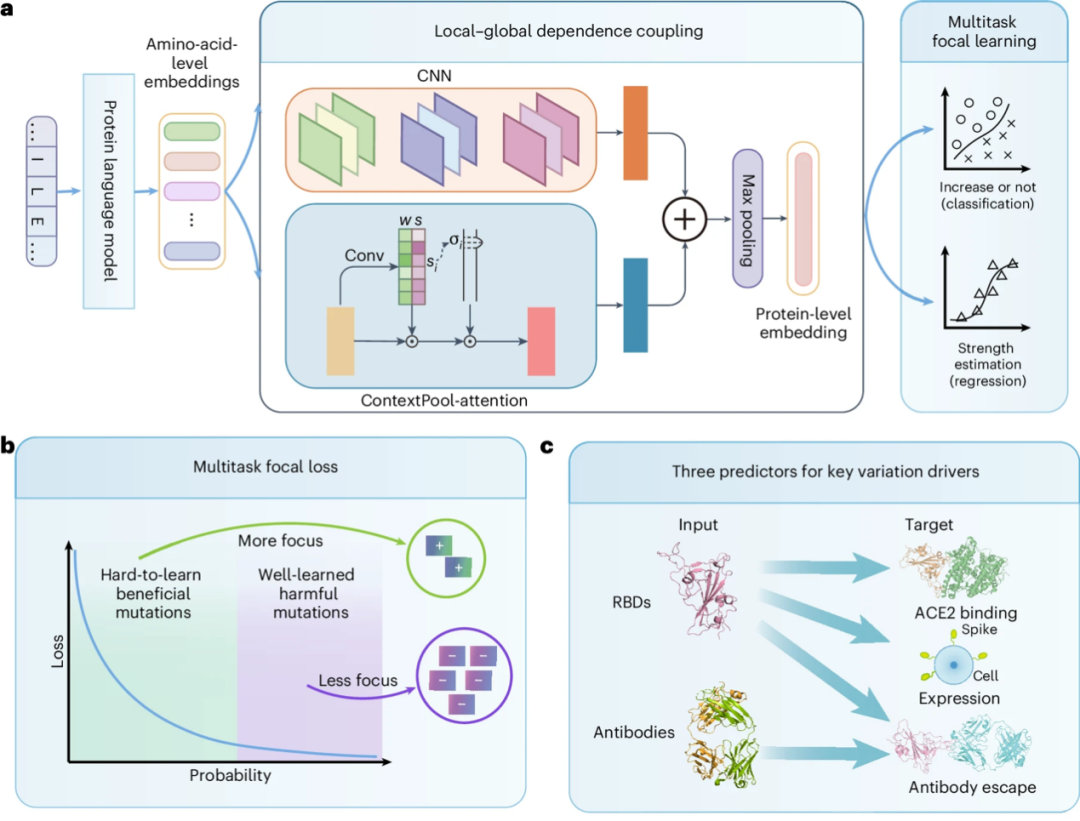
* d'abord,Dans le module de codage des séquences protéiques, l’équipe de recherche a formé de manière indépendante un modèle de langage protéique personnalisé pour l’évolution virale, qui peut extraire avec précision les caractéristiques des séquences protéiques virales ;
* Deuxièmement,Dans le module de fusion des dépendances d'interaction locales-globales, les chercheurs ont utilisé des réseaux neuronaux convolutifs (CNN) pour capturer les dépendances d'interaction entre les mutations et les acides aminés voisins, et ont conçu un mécanisme d'attention dynamique apprenable pour construire un réseau de dépendance d'interaction à longue portée au niveau du motif où se trouve la mutation. Cette conception résout efficacement le problème selon lequel les effets faibles causés par un nombre global de mutations moins élevé dans la variante sont difficiles à capturer ;
* Alors,Dans le module d'apprentissage multitâche, les avantages de l'apprentissage multitâche et des stratégies d'extraction d'échantillons difficiles sont combinés pour améliorer les performances prédictives du modèle en matière d'aptitude à la mutation virale grâce au partage des paramètres de l'entraînement multitâche.
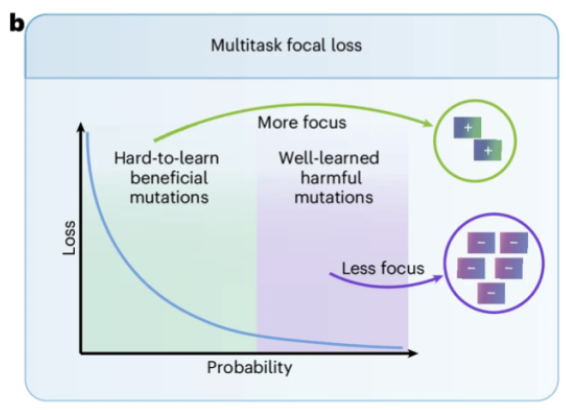
Plus important encore, comme le montre la figure b ci-dessus, l’équipe a conçu une nouvelle fonction de perte focale multitâche, qui incite le modèle à accorder plus d’attention aux mutations bénéfiques rares qui sont difficiles à apprendre efficacement pendant la formation, améliorant ainsi considérablement les performances de prédiction pour les mutations bénéfiques rares (c’est-à-dire les échantillons difficiles).
De plus, comme le montre la figure c ci-dessus, le cadre de prédiction E2VD peut ajuster de manière flexible l’entrée et la sortie pour diverses tâches de prédiction de la fitness du virus. Par exemple, pour prédire les changements d’affinité de liaison causés par des mutations, seule la séquence du virus peut être saisie ; pour prédire les changements dans la capacité d'échappement des anticorps causés par des mutations, la séquence du virus et la séquence de l'anticorps peuvent être saisies, etc., permettant ainsi d'obtenir des prédictions évolutives de haute précision sur les types et les souches de virus sur une architecture unifiée.
Plus précisément, dans l’étude, le cadre E2VD a été utilisé pour les tâches de prédiction liées au virus SARS-CoV-2, à la grippe (virus de la grippe), au virus Zika (virus Zika) et au VIH (virus du SIDA) :
* Les tâches du SARS-CoV-2 comprennent la prédiction de l’affinité de liaison, de l’expression et de l’échappement des anticorps, qui sont les principaux moteurs de la mutation du virus.
* La tâche pour les virus de la grippe, du Zika et du VIH est de prédire l’effet de fitness causé par les mutations afin d’analyser la capacité de généralisation du modèle.
Résultats expérimentaux : E2VD améliore la précision de la prédiction des mutations bénéfiques par 67% et présente d'excellentes performances de généralisation
E2VD peut capturer avec précision les modèles d'évolution virale et améliorer la précision de la prédiction des mutations bénéfiques par 67%
L’équipe a comparé les performances de prédiction du langage protéique personnalisé pour les scénarios évolutifs avec celles du modèle de langage protéique traditionnel. Les résultats ont montré que le modèle de langage protéique personnalisé de l'équipe a obtenu les meilleures performances de prédiction avec un minimum de 340 millions de paramètres de modèle, surpassant même l'ESM2-15B, qui a 44 fois plus de paramètres. Cela prouve l’efficacité de l’ensemble de données de pré-formation personnalisé et de la stratégie de formation.
Par la suite, l’équipe a comparé l’E2VD avec les méthodes traditionnelles dans diverses tâches clés de prédiction des forces motrices de l’évolution virale. Les résultats ont montré que l’E2VD surpassait considérablement les autres méthodes, avec des améliorations de performances allant de 7% à 21%. De plus, afin de démontrer la capacité de l'E2VD à capturer avec précision les modèles d'évolution virale, comme la distinction précise des différents types de mutations et l'extraction précise des mutations bénéfiques rares, les chercheurs ont mené plusieurs expériences.
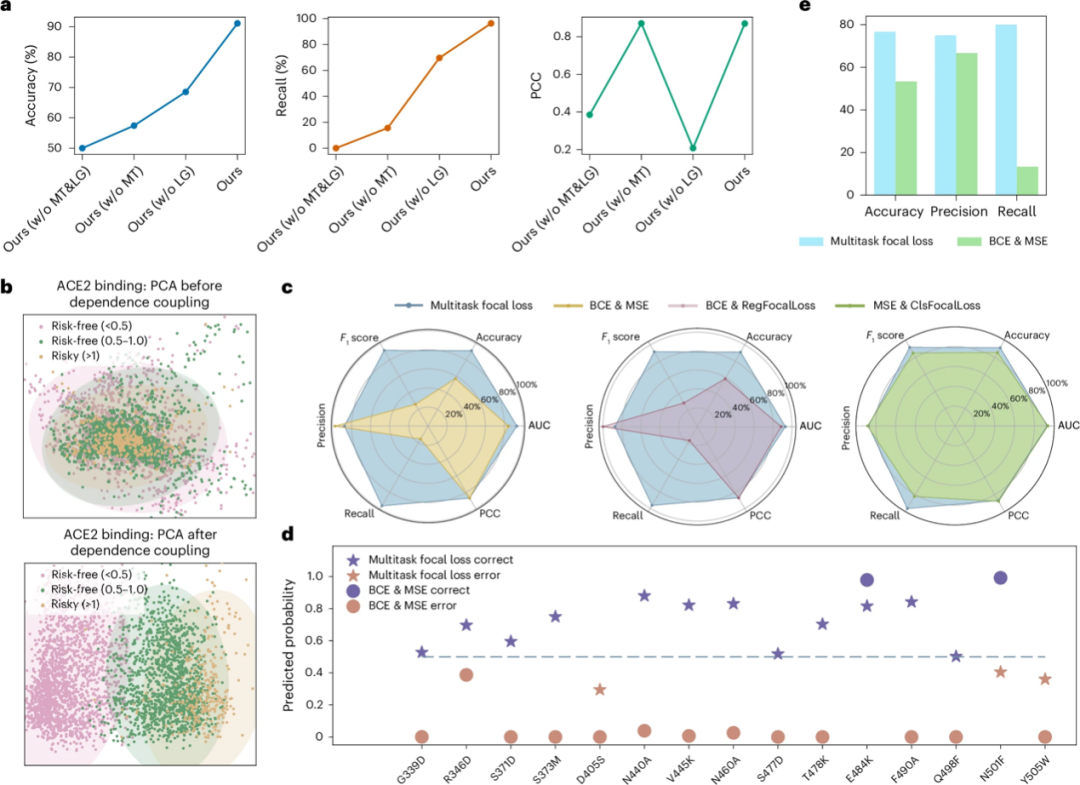
a : sans MT signifie E2VD sans module MT ; sans LG signifie E2VD sans module LG ; sans MT&LG signifie E2VD sans module MT&LG
b : Trois types de mutation avec des niveaux de risque décrits dans la tâche de prédiction d'affinité de liaison
d : La capacité de différentes pertes à capturer des mutations bénéfiques rares
Dans un premier temps, des études d’ablation de modules sont réalisées pour explorer les contributions du module de fusion de dépendance d’interaction locale-globale (LG) et du module d’apprentissage focal multitâche (MT) aux performances de prédiction. Comme le montre la figure a ci-dessus, l’étude a révélé que le module MT est efficace pour extraire les rares mutations bénéfiques dans la forme physique virale (le taux de rappel est passé de 0 à 69,63%). La combinaison du module LG avec le module MT peut encore améliorer les performances du modèle, avec une précision de 91,11%, un rappel de 96,3% et un coefficient de corrélation de 0,87.
La fonction de perte focale multitâche proposée par l’équipe peut améliorer considérablement les performances de prédiction. Pour évaluer la capacité de la perte focale multitâche à capturer des mutations bénéfiques rares, les chercheurs ont sélectionné des mutations bénéfiques et délétères représentatives pour former un ensemble de tests.
* En termes de prédiction de mutations bénéfiques, comme le montre la figure d ci-dessus, E2VD améliore la précision de prédiction des mutations bénéfiques rares de 13% à 80%, réalisant une amélioration considérable de la précision, permettant ainsi d'extraire avec précision et efficacité les mutations bénéfiques rares qui sont cruciales pour l'évolution virale.
* Pour les mutations nocives, la perte focale multitâche et le BCE&MSE traditionnel fonctionnent de manière similaire. Cela est dû au fait que BCE&MSE ne peut pas aider le modèle à apprendre les rares mutations bénéfiques, ce qui fait que le modèle a tendance à prédire toutes les mutations comme des mutations nuisibles.
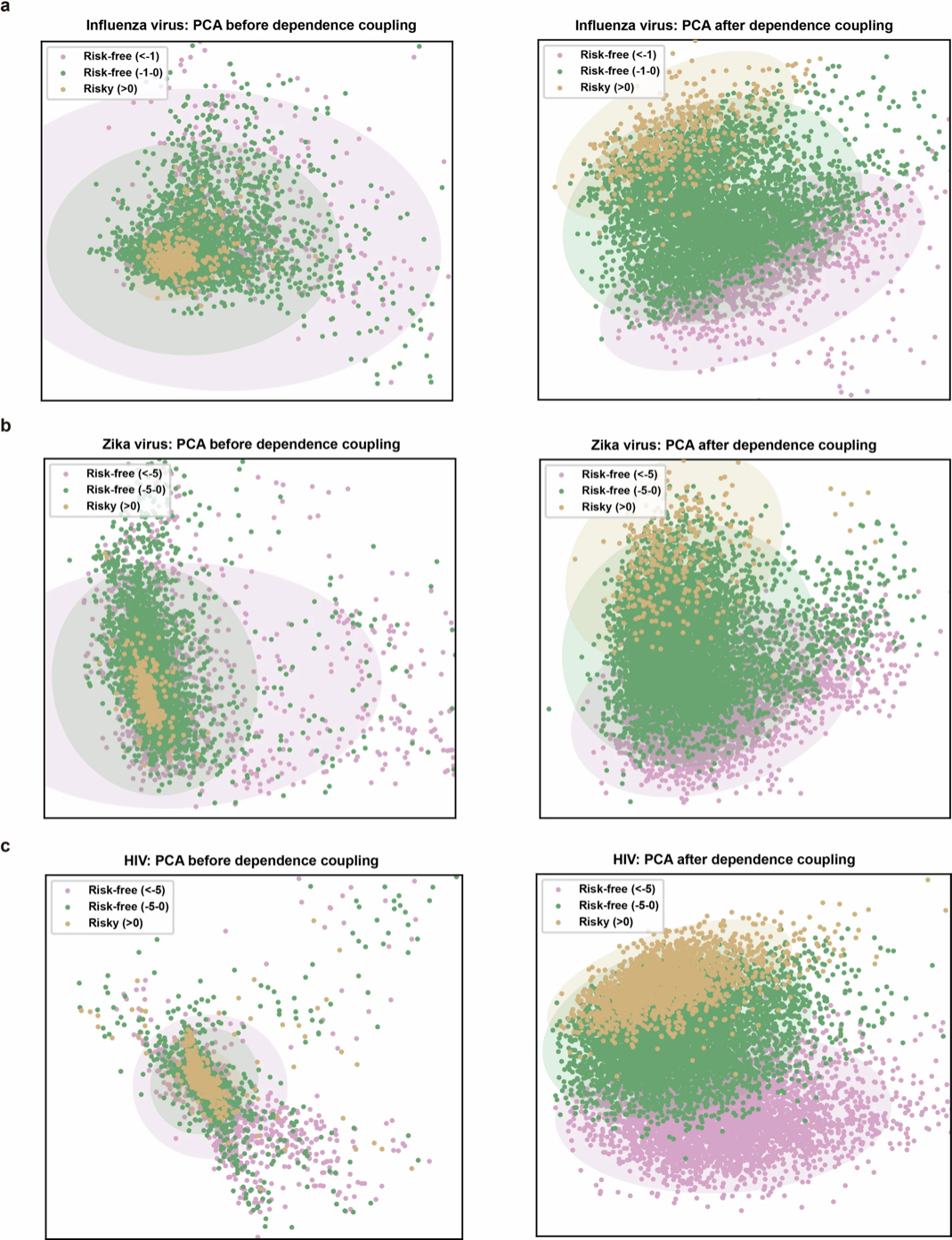
Comme le montre la figure b ci-dessous, les chercheurs ont utilisé l’analyse en composantes principales (ACP) pour visualiser la réduction de dimensionnalité de trois types de mutations dans la grippe, le virus Zika et le VIH. Les résultats ont montré qu’après traitement par le module LG, les caractéristiques des différentes mutations étaient clairement distinguées avec des limites claires. Cela suggère que LG peut améliorer la sensibilité de l'E2VD à divers types de mutations en capturant et en reconstruisant le réseau d'interaction intramoléculaire, offrant ainsi une meilleure compréhension de l'adaptabilité évolutive du virus.
L'E2VD présente d'excellentes performances de généralisation et peut faire des prédictions sur tous les types et souches de virus
Les virus continuent d’évoluer sous la pression de la sélection, ce qui peut conduire à l’émergence de multiples souches. Par exemple, le virus de la grippe qui a récemment suscité beaucoup d’attention comprend plusieurs types et présente des mutations saisonnières. Par conséquent, la capacité de généralisation du modèle est cruciale pour faire face aux tendances complexes de l’évolution des virus. Les chercheurs ont proposé la « proportion de paires ordinales » (OPP) pour évaluer la capacité de généralisation du modèle à prédire différentes souches du même virus et différents types de virus.
* OPP représente la proportion de paires de mutations correctement prédites parmi toutes les paires de mutations. Plus la valeur OPP est élevée, moins le paysage adaptatif prédit est chaotique, ce qui indique que le modèle est plus capable de prédire l’ordre relatif des facteurs de mutation virale.
Comme le montre la figure b ci-dessous, pour la tâche de prédiction de l'affinité de liaison inter-souches, les chercheurs ont évalué l'OPP de 6 souches différentes et de toutes les données mixtes de souches (toutes), et ont constaté que l'E2VD surpassait significativement les autres méthodes dans tous les cas. Comme le montre la figure c ci-dessous, E2VD surpasse les autres méthodes dans la tâche de prédiction du niveau d’expression pour la plupart des souches. Dans l’ensemble, l’E2VD surpasse largement les méthodes de pointe sur les souches hors distribution, démontrant des performances hautement généralisables.
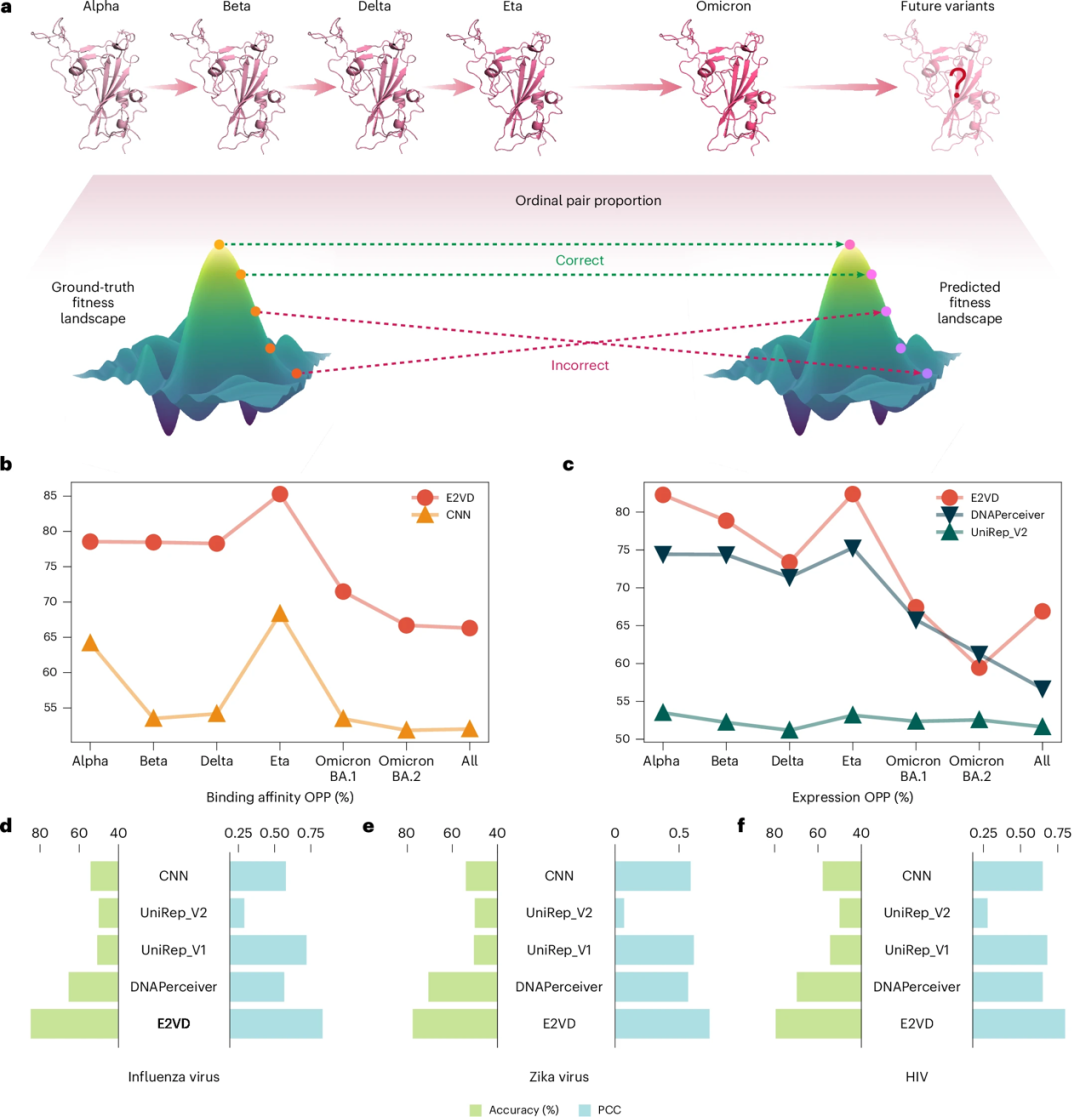
* b, c : E2VD prédit l’OPP de différentes souches virales ; d, e, f : E2VD prédit les performances de différents types de virus
Comme le montrent les figures d, e et f ci-dessus, dans les prédictions de types de virus croisés, les chercheurs ont découvert que l'E2VD présentait des capacités de généralisation idéales pour le nouveau coronavirus, le virus Zika, le virus de la grippe et le VIH, surpassant largement les autres méthodes, et pourrait être étendu à des virus plus infectieux à l'avenir.
L’IA a un grand potentiel pour prédire l’évolution virale
La recherche ci-dessus a réexploré le problème de la prédiction de l’évolution des virus du point de vue de l’évolution et a construit un cadre de prédiction de l’évolution universel E2VD adapté à différents types et souches de virus. Ce cadre a montré d’excellentes performances prédictives et une capacité de généralisation dans les tâches de prédiction de multiples facteurs moteurs de mutation virale, permettant de prédire les tendances d’évolution du virus.De plus, la combinaison flexible et personnalisée d’E2VD peut également réaliser la prédiction des tendances évolutives à différentes échelles.
* Premièrement, l’E2VD peut expliquer le cheminement de l’évolution virale au cours des pandémies, nous aidant à comprendre les raisons de la prévalence des souches et les mécanismes moléculaires qui les sous-tendent.
* Deuxièmement, combiné à une simulation de balayage de mutations profondes virtuelles, E2VD est capable de prédire d'éventuelles mutations à haut risque, atteignant un taux de réussite de 80%.
* Enfin, E2VD permet également de prédire la trajectoire macroévolutive à l’échelle de la pandémie, en reproduisant le chemin évolutif du virus dans le monde réel, fournissant ainsi un support théorique à l’interprétation du mécanisme d’évolution du virus.
À l’avenir, l’équipe prévoit de combiner l’E2VD avec les processus de conception de vaccins et de médicaments protéiques pour améliorer l’efficacité et la contrôlabilité de la conception, ce qui sera d’une grande importance pour la prévention et le contrôle viraux et la conception de médicaments.
Il convient de mentionner que les auteurs de l'étude sont le professeur Tian Yonghong et le professeur associé Chen Jie de l'École d'ingénierie de l'information de l'Université de Pékin, ainsi que leurs étudiants au doctorat Nie Zhiwei et l'étudiant en master Liu Xudong. L’équipe continue de se concentrer sur la recherche dans le domaine de l’IA pour les sciences de la vie. Leur projet « Ahead of the Evolution of the Virus - Predicting Future High-Risk Coronavirus Variants through Artificial Intelligence Simulation » a été présélectionné avec succès pour le « Gordon Bell New Crown Special Award » 2022 en novembre 2022 (le prix Gordon Bell est la plus haute distinction académique dans le domaine des applications de calcul haute performance au monde).
L’équipe possède une expérience approfondie dans le domaine de la prédiction de l’évolution des virus. En juillet 2023, l'équipe a publié « Running ahead of evolution—AI-based simulation for predicting future high-risk SARS-CoV-2 variants » dans The International Journal of High Performance Computing Applications. Plus précisément, les chercheurs ont pré-entraîné un grand modèle de langage protéique et construit une méthode de criblage à haut débit basée sur l’affinité de liaison et la prédiction de l’échappement des anticorps. Il s’agit de la première étude sur la simulation des mutations du RBD du SARS-CoV-2. Le modèle a identifié avec succès des mutations dans la région RBD de cinq variantes préoccupantes et a éliminé des millions de variantes potentielles en quelques secondes, fournissant un moyen technique de prévention et de contrôle des épidémies sous la forme d'un paradigme « IA+HPC » (intelligence artificielle + calcul haute performance).
Lien vers l'article :
https://journals.sagepub.com/doi/abs/10.1177/10943420231188077
En outre, l’équipe a développé une série de modèles de base pour les sciences de la vie. Prenant comme exemple la tâche de prédiction de l'interaction « enzyme-substrat », qui est cruciale pour l'ingénierie enzymatique, l'équipe a publié un article préliminaire en décembre 2024, proposant un cadre d'apprentissage profond conditionnel progressif MESI pour la prédiction polyvalente de l'interaction enzyme-substrat.
Lien vers l'article :
https://www.researchsquare.com/article/rs-5516445/v1
Plus précisément, en découplant la modélisation des interactions enzyme-substrat en un processus d'apprentissage en deux étapes, deux réseaux conditionnels sont conçus pour introduire respectivement la spécificité de la réaction enzymatique et les informations clés sur l'interaction catalytique, facilitant ainsi la transition progressive de l'espace latent des caractéristiques du domaine général des protéines et des petites molécules au domaine conscient de la catalyse. Le modèle surpasse systématiquement les méthodes de pointe sur diverses tâches en aval. De plus, le réseau conditionnel proposé capture implicitement les modes essentiels de la catalyse enzymatique avec une surcharge de calcul supplémentaire négligeable. Grâce à ce mécanisme de détection conditionnelle, le modèle peut identifier avec précision les sites actifs et explorer les résidus d'enzymes et les groupes fonctionnels de substrat impliqués dans les interactions catalytiques clés de manière efficace et peu coûteuse sans avoir besoin d'informations structurelles.
Avec l'aide de l'intelligence artificielle, l'équipe favorisera davantage la recherche approfondie dans les domaines connexes de l'IA pour les sciences de la vie, ouvrira davantage de possibilités pour la prédiction des virus, la conception de médicaments protéiques, le développement de vaccins, etc. Nous attendons avec impatience d'autres de leurs réalisations.
Références :
https://www.who.int/
https://news.pku.edu.cn/jxky/90d276ae5f8441849fd04372fd872154.htm
https://news.pkusz.edu.cn/info/1003/8711.htm









