Command Palette
Search for a command to run...
Professeur Zheng Wei De l'Université De Nankai : AlphaFold n'est Pas Parfait, Et La Communauté Universitaire a Encore La Possibilité De « Prendre Le Dessus »

Ces dernières années, avec l’aide des technologies d’IA telles que l’apprentissage profond, le domaine de la prédiction de la structure des protéines s’est développé rapidement. En octobre 2024, Demis Hassabis et John M. Jumper de DeepMind ont remporté le prix Nobel de chimie 2024 grâce à AlphaFold. Cependant, cela ne signifie pas qu’AlphaFold est irremplaçable, et d’autres excellents algorithmes méritent toujours d’être explorés.
Dans le sixième épisode de la série en direct « Meet AI4S »,HyperAI a l'honneur d'avoir invité le professeur Zheng Wei, professeur à l'École de statistique et de science des données de l'Université de Nankai,Sous le thème « Le trône d'AlphaFold3 n'est pas stable et la communauté universitaire le dépasse : prédiction de la structure tridimensionnelle des macromolécules biologiques et de leurs interactions basée sur l'apprentissage profond », il a partagé avec tout le monde les limites d'AlphaFold et les futures directions d'optimisation, ainsi que les algorithmes et les sujets de recherche qui méritent d'être explorés dans la communauté universitaire.
* Suivez le compte officiel et répondez « Meet AI4S 6th » pour obtenir la présentation PPT
HyperAI a organisé et résumé le partage approfondi sans violer l'intention initiale. Voici la transcription du discours.
Limites d'AlphaFold
Les protéines sont les pierres angulaires des activités de la vie, et la prédiction de leurs structures tridimensionnelles est essentielle à la compréhension de leurs fonctions biologiques. Bien qu'AlphaFold 2 lancé par DeepMind ait porté la prédiction de la structure des protéines à un nouveau niveau, cela ne signifie pas que le cadre de bout en bout d'AlphaFold 2 a résolu tous les problèmes de prédiction de la structure des protéines.
Tout d’abord, en prenant AlphaFold 2 lui-même comme exemple, il présente encore de nombreuses limitations :
* La précision doit être améliorée
Les rapports officiels montrent qu'AlphaFold 2 peut prédire les structures avec une précision de plus de 90%, mais la tâche réelle ne peut pas atteindre un niveau aussi élevé.
* La prédiction de la structure des protéines multi-domaines est limitée
AlphaFold 2 est performant dans la prédiction des protéines à domaine unique, mais pour les protéines multi-domaines complexes, où les domaines sont relativement flexibles, la précision de la prédiction n'est pas bonne.
* La prédiction de la structure des complexes protéiques est limitée
Les protéines doivent généralement former des complexes avec d’autres protéines pour fonctionner, mais la version initiale d’AlphaFold 2 n’a pas résolu ce problème.
* La prédiction de la structure de l'ARN, la prédiction de la structure ARN-ARN et protéine-ARN sont limitées
Comme indiqué ci-dessus, ces problèmes n’ont pas été abordés dans la version initiale.
* La prédiction de la dynamique des protéines/des changements conformationnels est limitée
Les méthodes d’analyse expérimentale ne peuvent généralement capturer que l’état structurel à un certain moment, mais les protéines n’existent pas de manière statique dans leurs organismes et leurs structures à différents moments peuvent être différentes. Ces problèmes n’ont pas encore été résolus par AlphaFold 2.
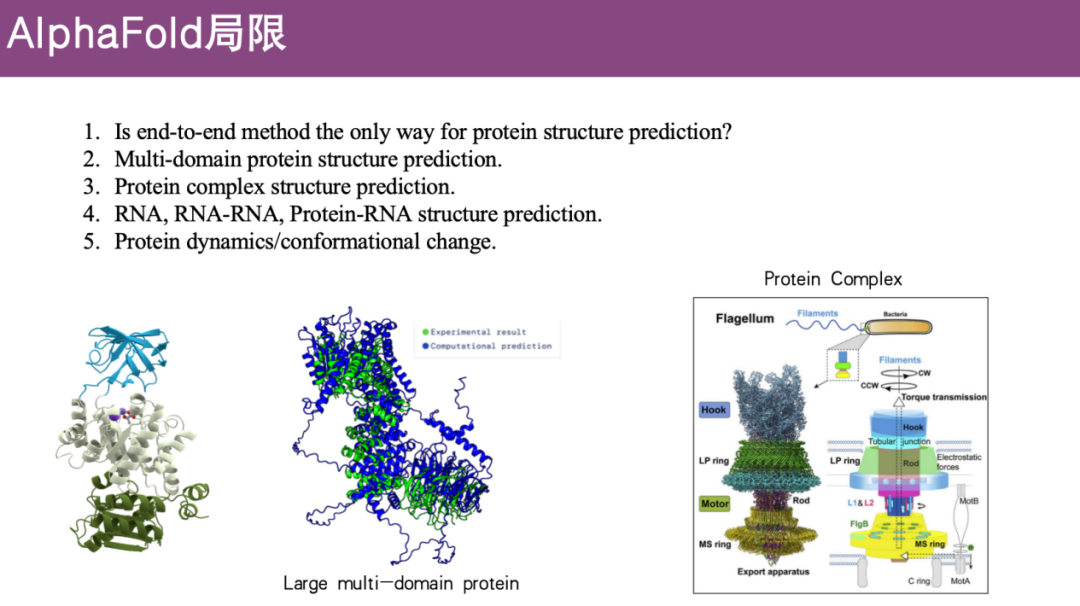
De plus, bien que DeepMind ait itéré AlphaFold 3, et que nous sachions tous qu'il est performant dans la prédiction des structures des monomères protéiques, sa précision dans la prédiction des complexes, des acides nucléiques et des petites molécules doit encore être améliorée. donc,La prochaine génération d'AlphaFold pourrait ajouter des modules de prédiction avec d'autres fonctions.Par exemple, étant donné que les modèles existants sont principalement utilisés pour traiter des structures statiques, nous explorerons les processus de dynamique moléculaire et prédirons les conformations des protéines. En outre, cela pourrait également impliquer le domaine de la conception des protéines, inversant ainsi l’ensemble du processus de prédiction.
Par conséquent, même avec AlphaFold, il reste encore beaucoup de travail à faire dans le monde universitaire.
En plus d’AlphaFold, existe-t-il d’autres méthodes qui méritent d’être explorées ?
Dans le passé, les principales méthodes que nous utilisions pour résoudre la structure tridimensionnelle des protéines étaient les rayons X, la résonance magnétique nucléaire (RMN) et la cryomicroscopie électronique. En raison de la difficulté et du coût élevé de la résolution expérimentale des structures protéiques, certaines équipes peuvent avoir besoin de passer des mois, voire des années, pour résoudre la structure tridimensionnelle d’une protéine. En conséquence, les gens ont commencé à explorer une méthode plus économique et plus rapide, qui consiste à prédire la structure des protéines grâce à des algorithmes.
Nous savons que les protéines sont principalement composées de 20 types d’acides aminés, généralement représentés par des lettres anglaises, et que les molécules d’acides aminés contiennent également de nombreux atomes.Par conséquent, le problème de prédiction de la structure des protéines peut être résumé comme suit : entrez une chaîne d'acides aminés composée de ces lettres et utilisez un algorithme de calcul pour prédire les coordonnées spatiales tridimensionnelles (x, y, z) de chaque atome dans chaque acide aminé de la séquence protéique.
En examinant l'ensemble de l'historique du développement de la prédiction de la structure des protéines, une variété d'algorithmes représentatifs ont émergé à différentes étapes, tels que la modélisation comparative ou la modélisation d'homologie, la simulation de dynamique moléculaire (MD), l'algorithme de threading, la prédiction de novo et l'algorithme de prédiction de structure basé sur la prédiction d'apprentissage profond des cartes de contact. L'introduction principale est la suivante :
* Modélisation comparative ou modélisation d'homologie
Cette méthode est basée sur les principes de l’évolution biologique.On pense que si la similarité de séquence est élevée, la structure et la fonction de la protéine seront également relativement similaires.Par conséquent, nous pouvons d'abord obtenir la séquence d'acides aminés de la protéine inconnue, puis trouver le modèle de structure protéique résolu avec une similarité de séquence élevée dans la base de données PDB grâce à l'alignement de séquence, et prédire la structure de la protéine inconnue grâce à la migration ou à l'alignement.
*La base de données PDB contient les structures des protéines qui ont été résolues dans ce domaine

* Simulation de dynamique moléculaire
L'idée de base est de générer aléatoirement une structure tridimensionnelle initiale basée sur la séquence d'acides aminés de la protéine, d'attribuer des coordonnées aléatoires à chaque atome, d'ajuster la position atomique, puis de calculer l'énergie d'état de la protéine à différents moments en fonction du champ d'énergie physique pré-construit.La structure avec l’énergie la plus faible est la conformation protéique raisonnable.

* Algorithme de threading
Similaire à la modélisation d'homologie, la différence est que, bien que les protéines présentant une similarité de séquence élevée soient souvent similaires en termes de structure, les protéines présentant des structures similaires peuvent avoir une faible similarité de séquence, et ces protéines ne peuvent pas trouver d'informations de modèle appropriées dans la base de données PDB. Les chercheurs ont ensuite proposé le concept de profil et, sur la base des séquences homologues collectées, ont utilisé l'alignement de séquences multiples (MSA) pour aligner différents acides aminés de la même manière que l'on aligne deux profils protéiques.
C'est-à-dire, même si les deux séquences d'acides aminés sont différentes,Mais leurs profils sont similaires, nous pouvons donc supposer que leurs structures sont similaires.Utilisez ceci pour trouver des modèles.

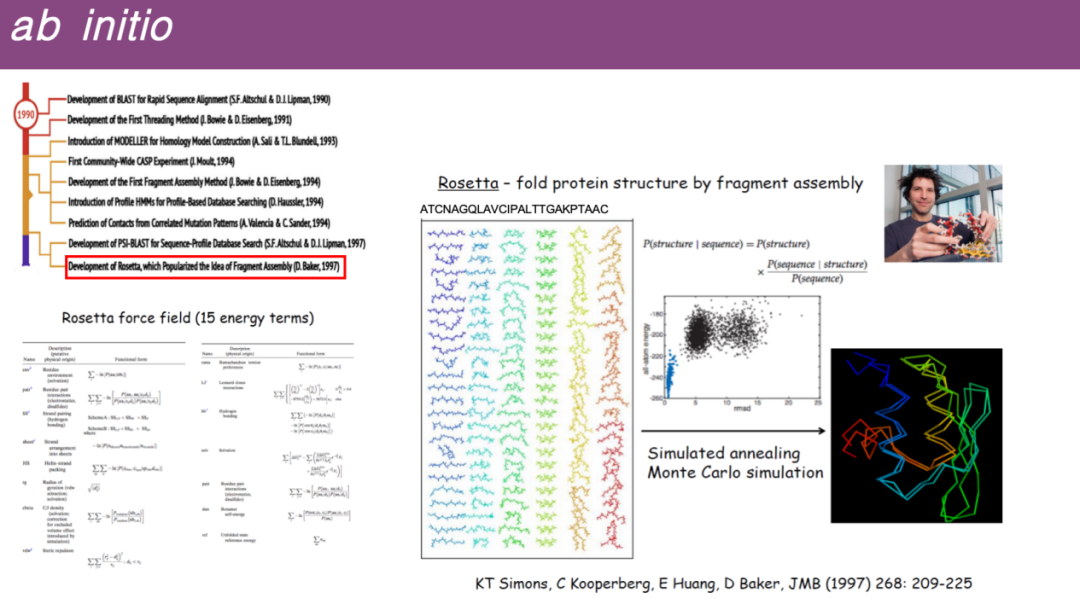
* Prédiction de novo
Certaines protéines peuvent ne pas avoir de structures similaires dans la base de données.Les chercheurs ont ensuite essayé de faire des prédictions en décomposant la séquence protéique entière en fragments plus courts, en recherchant des modèles de ces petits fragments dans la base de données, puis en assemblant ces modèles de petits fragments dans une structure tridimensionnelle complète.
Plus précisément, le professeur David Baker de l'Université de Washington a développé le logiciel Rosetta, dont le principe principal est de décomposer la séquence protéique en de nombreux petits fragments, d'assembler aléatoirement ces fragments, puis de les optimiser en utilisant la fonction énergétique développée dans la simulation de dynamique moléculaire, et d'effectuer une prédiction de structure grâce à des principes similaires à la simulation dynamique et à la minimisation de l'énergie.
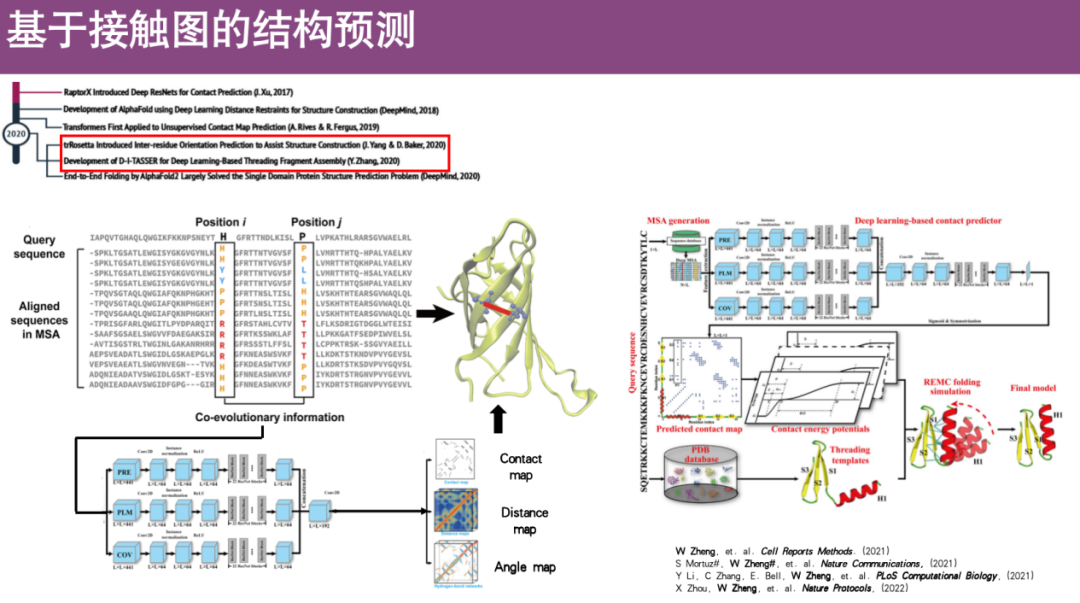
* Plan de contact
L’idée principale est de convertir la structure tridimensionnelle d’une protéine en un graphique bidimensionnel.Les informations sur la structure tridimensionnelle des protéines sont utilisées, c'est-à-dire que les positions des coordonnées de tous les points spatiaux sont utilisées pour calculer la distance entre les différents acides aminés. On suppose qu'un contact se forme lorsque la distance entre deux acides aminés est inférieure à un certain seuil, sinon aucun contact ne se forme. Cette définition est utilisée pour compresser la structure tridimensionnelle dans un graphique bidimensionnel. De plus, les informations de cette carte de contact bidimensionnelle peuvent être utilisées pour reconstruire la structure tridimensionnelle de la protéine.
Plus précisément, les chercheurs ont développé de nombreuses méthodes basées sur l’apprentissage profond. L'idée principale est de construire d'abord un alignement de séquences multiples (MSA) pour observer les informations co-évolutives des profils dans les acides aminés i et j, car ces acides aminés co-évolutifs sont souvent très proches dans l'espace et formeront des contacts. Par la suite, les informations de coévolution sont entrées sous forme de caractéristiques dans le réseau d'apprentissage profond pour la formation, prédisant ainsi la carte de contact des protéines et restaurant la structure tridimensionnelle des protéines.
Par exemple, l’équipe du professeur Zheng Wei a précédemment développé un algorithme appelé CI-TASSER, qui est actuellement une méthode couramment utilisée pour prédire la structure des protéines sur la base de cartes de contact.
Enfin, AlphaFold intègre les principes de base de nombreux algorithmes ci-dessus et construit avec succès un cadre de bout en bout qui peut saisir directement des séquences de protéines, puis générer des structures.
Prenons l’exemple des réalisations d’équipe et explorons les opportunités pour les universitaires de se dépasser.
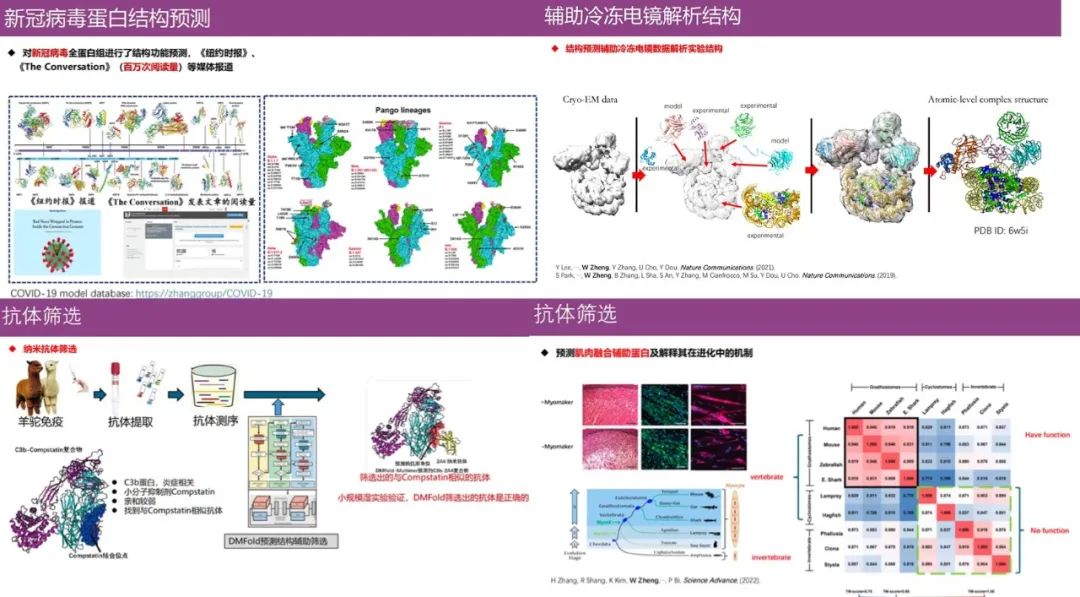
La prédiction de la structure des protéines a un impact énorme sur le domaine biomédical, par exemple,Les algorithmes actuellement développés par l'équipe du professeur Zheng Wei consistent à prédire les structures protéiques virales inconnues (nouveau coronavirus), à aider à l'analyse des structures protéiques à l'aide de la microscopie cryoélectronique, à aider les biologistes à comprendre les fonctions évolutives des protéines et à effectuer le dépistage des anticorps.
De plus, comme le montre la figure ci-dessous, tous les algorithmes de prédiction de structures monomères et complexes de protéines développés par l'équipe ont été convertis en algorithmes de serveur automatiques et publiés sur le site Web du groupe de recherche. Ses algorithmes ont servi plus de 90 000 utilisateurs dans plus de 100 pays à travers le monde, et tout le monde est invité à les utiliser.
*Adresse totale du projet :
https://seq2fun.dcmb.med.umich.edu/DMFold

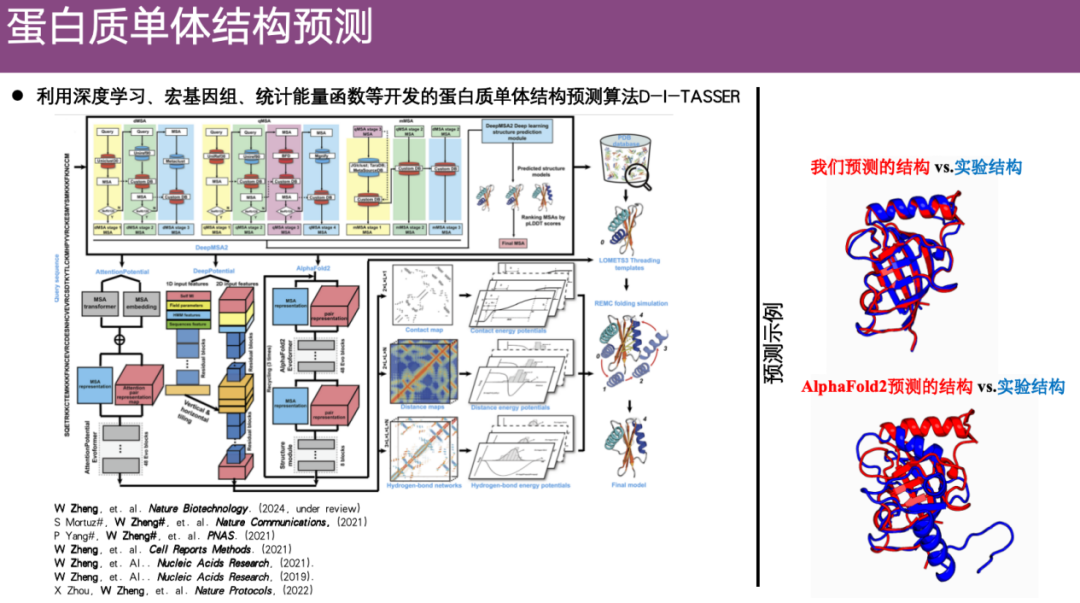
Méthode de prédiction de la structure des monomères protéiques DI-TASSER
Le problème de la prédiction de la structure des monomères protéiques a toujours attiré beaucoup d’attention. Avant AlphaFold 2, l’équipe du professeur Zheng Wei menait des recherches sur la prédiction de structure basées sur des cartes de contact. Après l’émergence d’AlphaFold 2, l’équipe a commencé à se demander si elle pouvait intégrer des contraintes spatiales telles que la carte de contact prédite par AlphaFold 2 dans les algorithmes précédemment développés. Ainsi, sur la base de contraintes spatiales, de métagénomes, de fonctions énergétiques statistiques, etc.L'équipe a développé un algorithme de prédiction de la structure des monomères protéiques DI-TASSER, qui a montré de bons résultats après optimisation.
Comme le montre l'exemple sur le côté droit de la figure ci-dessous, le rouge représente la structure protéique prédite par DI-TASSER et le bleu représente la structure analysée expérimentalement. Comme vous pouvez le voir,La structure prédite par DI-TASSER est très similaire à la structure résolue expérimentalement.En revanche, la structure prédite par AlphaFold 2 est significativement différente de la structure expérimentale même après alignement, et sa précision de prédiction est légèrement inférieure.
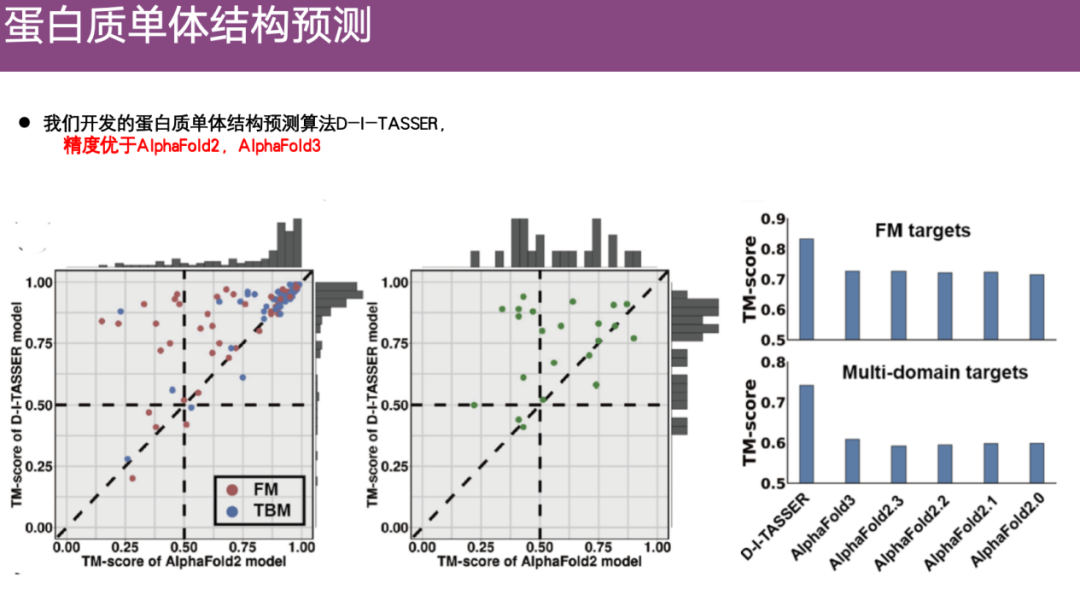
De plus, il est évalué sur plusieurs ensembles de données protéiques. Comme le montre le côté droit de la figure ci-dessous, lors de la prédiction d'un domaine unique et de plusieurs domaines,La précision de prédiction de DI-TASSER est supérieure à celle d'AlphaFold 2 et même supérieure à celle d'AlphaFold 3.
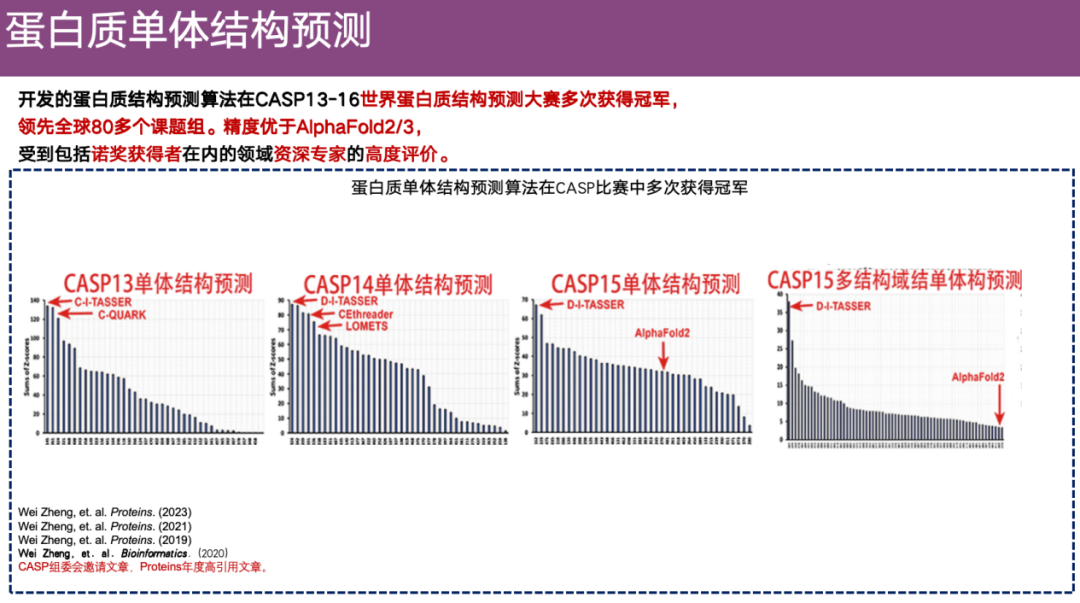
Pour garantir l'autorité de l'évaluation, l'équipe a non seulement mené des évaluations internes, mais a également participé au concours faisant autorité dans le domaine - CASP.
La compétition CASP est connue comme les Jeux olympiques dans le domaine et vise principalement à normaliser les méthodes d'évaluation pour la prédiction de la structure des protéines. Étant donné qu’il existe de nombreux types d’algorithmes de prédiction de la structure tridimensionnelle des protéines, chaque laboratoire a également développé son propre algorithme. Étant donné que les ensembles de données et les méthodes d’évaluation peuvent être différents, chaque groupe de recherche affirme généralement que sa méthode est la plus précise au monde. Pour résoudre ce problème, le concours CASP a été créé.
Depuis l'année dernière, le CASP a été organisé avec succès lors de 16 sessions et dure depuis 32 ans, attirant de nombreuses équipes faisant autorité, telles que l'équipe du professeur David Baker et l'équipe DeepMind.
DI-TASSER et ses algorithmes prédécesseurs ont participé à de nombreuses reprises aux compétitions CASP. Au cours des CASP 13 à 15, cette méthode a occupé une position de leader dans le domaine de la prédiction de la structure des monomères protéiques. Dans CASP 15,L’algorithme DI-TASSER a également participé à l’évaluation multi-domaines et sa précision globale était meilleure que celle de tous les groupes de recherche participants.
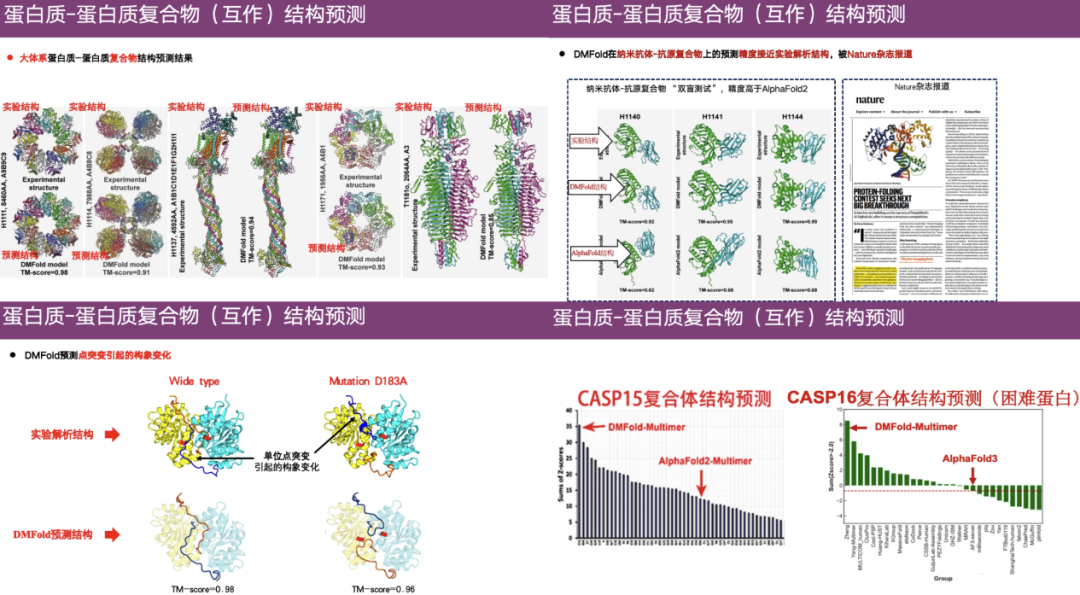
DMFold, une méthode de prédiction de la structure des complexes protéiques
Le principal défi dans la prédiction de structures complexes est de prédire la torsion relative entre deux protéines, qui peut être analysée à l’aide d’informations co-évolutives.
Par exemple, en construisant un alignement de séquences multiples (MSA) des monomères de deux protéines, en fusionnant les deux MSA en un seul MSA sur la base de certaines méthodes de connexion et en utilisant la relation co-évolutive des acides aminés entre les deux MSA pour déduire la distance entre les acides aminés dans différentes protéines, les informations co-évolutives peuvent également être intégrées dans le cadre d'apprentissage profond pour prédire la torsion relative entre deux protéines.
À cet égard,Le groupe de recherche du professeur Zheng Wei a développé les algorithmes DeepMSA et MetaSource pour construire des alignements de séquences multiples plus profonds.De plus, l’équipe a également utilisé l’apprentissage profond, la métagénomique, etc. pour développer un algorithme de prédiction de structure de complexe protéique DMFold.
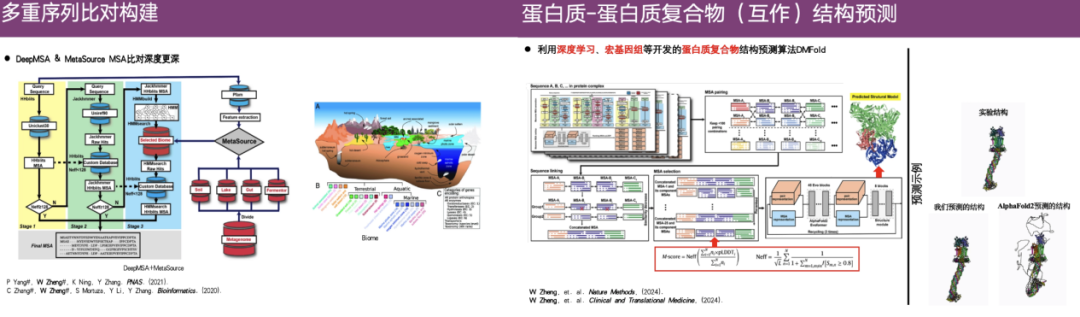
Comme le montre le cas à l'extrême droite de la figure ci-dessus, le haut est la véritable structure obtenue par analyse expérimentale, le bas à gauche est la structure prédite par DMFold et la droite est le résultat prédit par AlphaFold 2. On peut voir que la structure prédite par AlphaFold 2 est relativement chaotique et présente des extensions anormales en forme de tentacules. En revanche, la structure prédite de DMFold est très similaire à la structure expérimentale.Cela montre que l'algorithme DMFold est supérieur à AlphaFold 2 dans la prédiction de structures complexes.
De plus, DMFold montre également une grande précision dans les complexes protéine-protéine de grands systèmes, les complexes nanoanticorps-antigène, les changements conformationnels causés par des mutations ponctuelles, etc. Dans le cadre du concours CASP 15, Le classement général de DMFold est bien supérieur à celui d'AlphaFold 2, et dans CASP 16, DMFold est également meilleur qu'AlphaFold 3.
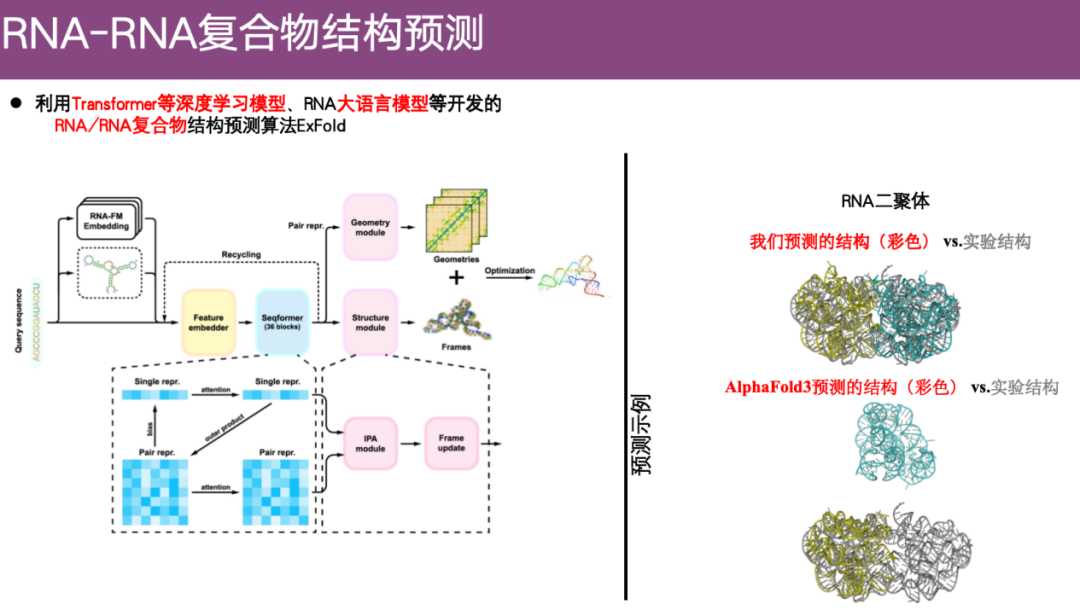
Méthode de prédiction de la structure du complexe ARN-ARN ExFold
Ces dernières années, l’équipe a commencé à se concentrer sur le problème de la prédiction de la structure de l’ARN. Par exemple, ils ont développé l’algorithme de prédiction de structure complexe ARN/ARN ExFold en utilisant des modèles d’apprentissage profond tels que Transformer et les modèles de langage volumineux ARN.
Comme le montre l’exemple sur le côté droit de la figure ci-dessous, la partie grise est la structure expérimentale et la partie colorée est la structure prédite. Comme vous pouvez le voir,En utilisant la méthode ExFold, les deux structures ont été bien alignées. En revanche, la prédiction d’AlphaFold 3 a montré qu’il n’y avait même pas de contact entre les deux molécules d’ARN, ce qui peut presque être considéré comme complètement faux.
L’équipe a également comparé la précision d’ExFold 3 avec celle d’AlphaFold 3 en utilisant un ensemble de données plus vaste, comme indiqué sur le côté gauche de la figure ci-dessous. L'axe Y représente la précision de prédiction d'ExFold.L'axe des X représente la précision de prédiction d'AlphaFold 3. On peut voir que les avantages d'ExFold sont toujours assez évidents.
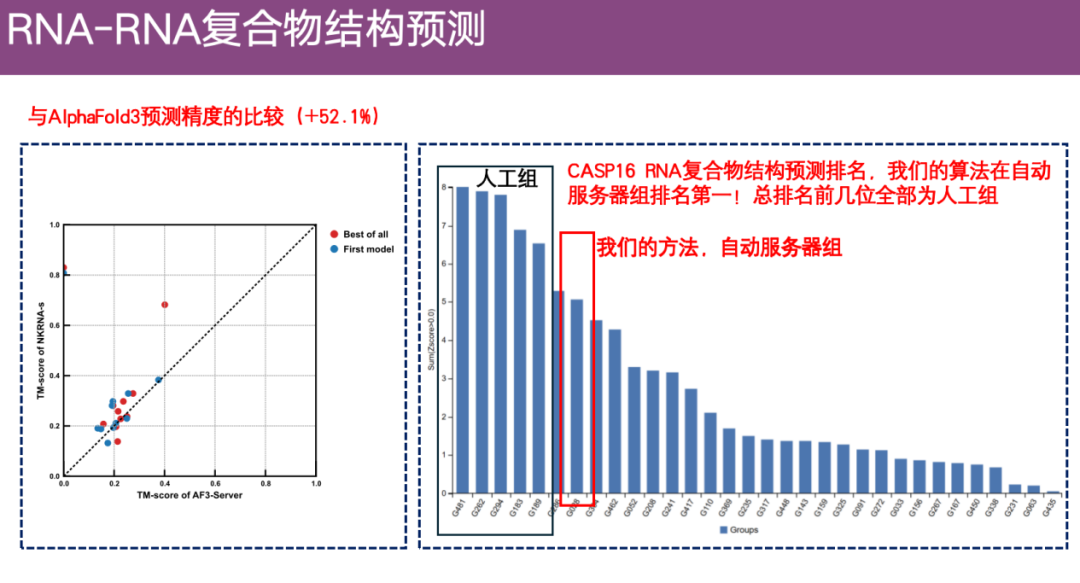
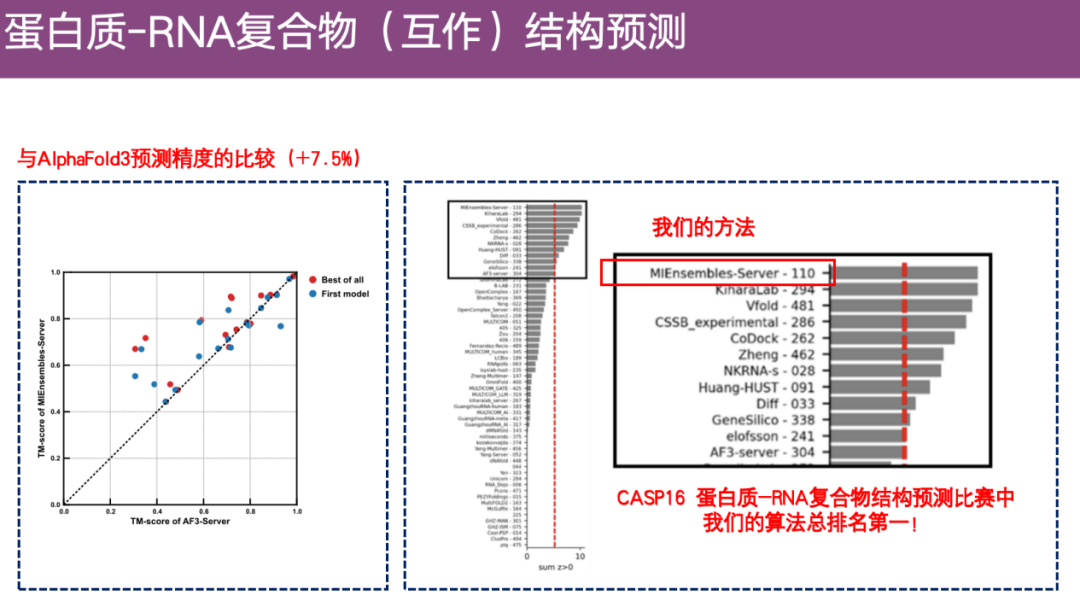
De plus, dans le cadre du concours de prédiction de la structure du complexe d'ARN CASP 16,Bien qu'ExFold ne soit pas classé premier au classement général, il se classe au premier rang parmi tous les algorithmes automatiques (algorithmes de serveur).
* La compétition CASP est divisée en groupe automatique et groupe manuel. Le groupe automatique est tenu de soumettre les résultats de prédiction de manière entièrement automatique dans un délai de 3 jours, et aucune intervention humaine n'est autorisée ; le groupe manuel a 3 semaines et est autorisé à ajouter une expérience experte et des ajustements manuels.
Méthode de prédiction de la structure du complexe protéine-ARN DeepProtNA
Concernant le problème de la prédiction de la structure du complexe protéine-ARN, l'équipe a utilisé des modèles d'apprentissage en profondeur tels que Transformer et le modèle de langage large récemment populaire pour développer un nouvel algorithme de prédiction de structure - DeepProtNA.
Comme le montre l'exemple ci-dessous à droite, dans le complexe anticorps-ARN, les couleurs représentent les résultats de prédiction de DeepProtNA, tandis que le gris représente la structure expérimentale. Après l'alignement, nous pouvons constater queLa structure prédite de DeepProtNA est très cohérente avec la structure expérimentale (chevauchement gris et couleur).En particulier à l'interface entre la protéine d'anticorps et l'ARN antigène, la précision de prédiction est très élevée. En revanche, la structure prédite d’AlphaFold 3 ne chevauche guère la structure expérimentale et l’effet de prédiction est faible.
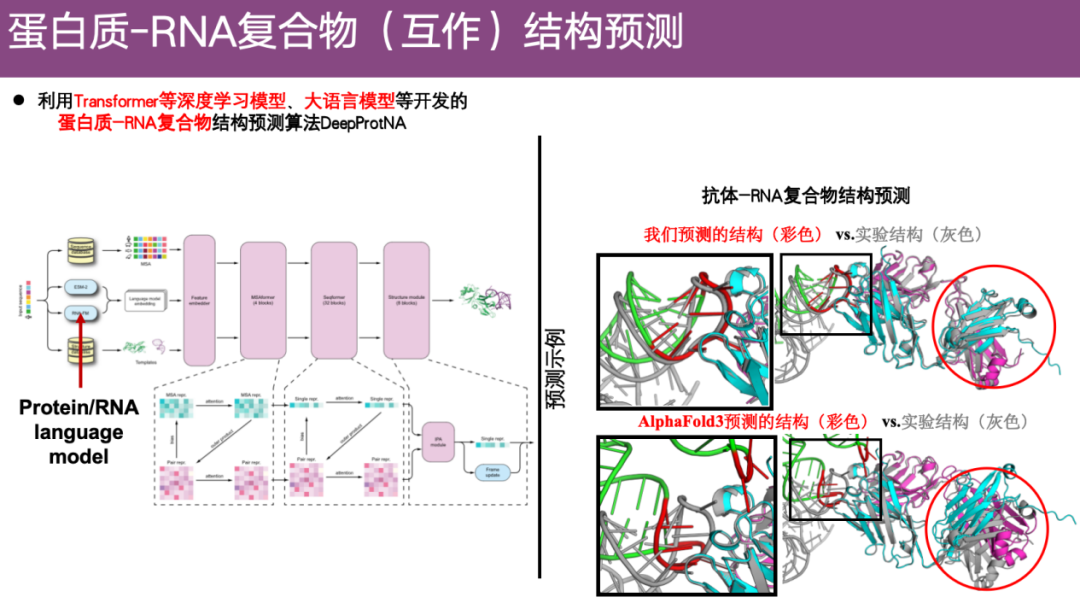
aussi,DeepProtNA est environ 7,5 points de pourcentage plus précis qu'AlphaFold 3.Classé premier dans la compétition du groupe serveur du CASP 16.
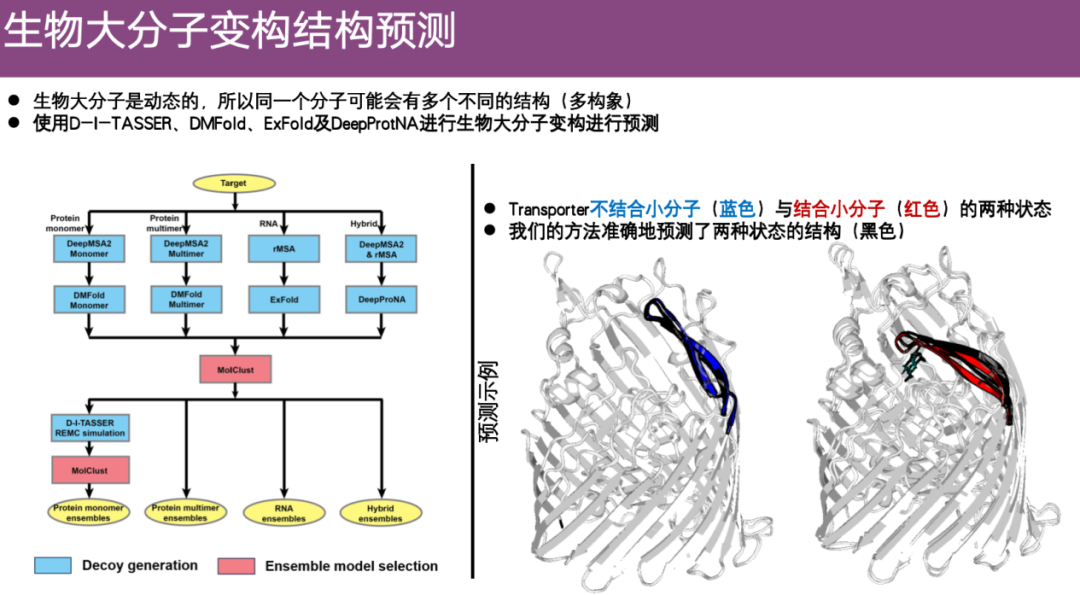
EnsembleFold : une méthode de prédiction des structures allostériques des biomacromolécules
L’équipe se concentre également sur le problème de la prédiction de la structure allostérique des macromolécules biologiques. L'entrée du problème de multi-conformation macromoléculaire est une séquence protéique et la sortie est constituée de plusieurs images clés de la protéine dans différents états. Cela signifie que, par rapport aux algorithmes de prédiction statique, plusieurs structures différentes doivent être prédites à partir d'une seule séquence d'acides aminés, et ces structures représentent les images clés de l'ensemble du processus dynamique. Il s’agit d’un sujet qui a reçu beaucoup d’attention dans le domaine actuel, mais qui est difficile à prévoir.
En intégrant des méthodes précédemment développées et en les optimisant pour l'allostérie macromoléculaire,L'équipe de recherche a développé des algorithmes de clustering et a finalement formé un algorithme appelé EnsembleFold.
Comme le montre l'exemple sur le côté droit de la figure ci-dessous, les changements conformationnels de la protéine après la liaison à la petite molécule sont démontrés. Le bleu représente la structure expérimentale lorsque la petite molécule n'est pas liée, et le rouge représente l'inclinaison et le changement de conformation après la liaison à la petite molécule verte. L'équipe a prédit deux structures basées sur la séquence protéique d'entrée, qui sont les parties noires. On peut voir que la structure prédite d’EnsembleFold est très cohérente avec la structure réelle lorsqu’elle n’est pas liée à de petites molécules. Après liaison à de petites molécules, EnsembleFold peut également bien s'adapter à la structure expérimentale. donc,EnsembleFold démontre une précision extrêmement élevée dans la prédiction des changements conformationnels dans les biomacromolécules.
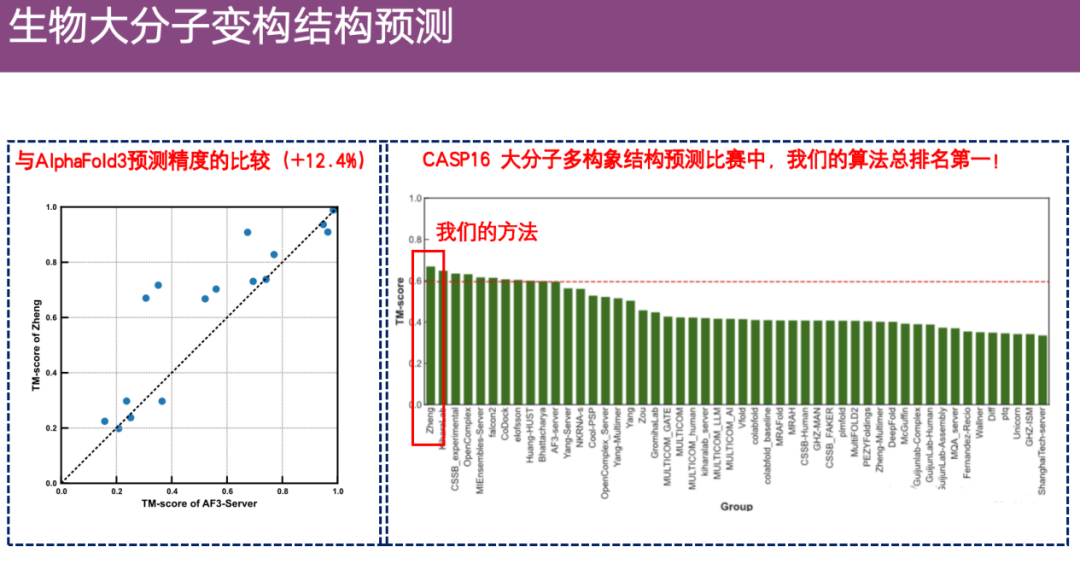
en même temps,Après comparaison avec AlphaFold 3, il a été constaté que la précision d’EnsembleFold était environ 12,4% supérieure.Il se classe au premier rang parmi toutes les compétitions de conformation macromoléculaire du CASP 16.
Un exemple intéressant est la prédiction par l’équipe des changements conformationnels de l’intégrase de l’ADN du bactériophage dans le CASP. Comme le montre la figure ci-dessous, la séquence d'acides aminés du bactériophage est représentée par P-P', et la séquence du matériel génétique de la bactérie est représentée par B-B'. Par un processus dynamique, l'intégrase de l'ADN du bactériophage intègre le matériel génétique P' du phage dans le matériel génétique B de la bactérie pour former B-P', et la conformation change.
L’équipe a utilisé des algorithmes pour prédire ces multiples changements conformationnels. Les structures expérimentales sont présentées à gauche, avec l'état non intégré (conformation 1) en haut et l'état intégré (conformation 2) en bas. On peut constater que les prédictions de l’équipe de recherche peuvent refléter avec précision ces deux conformations différentes.
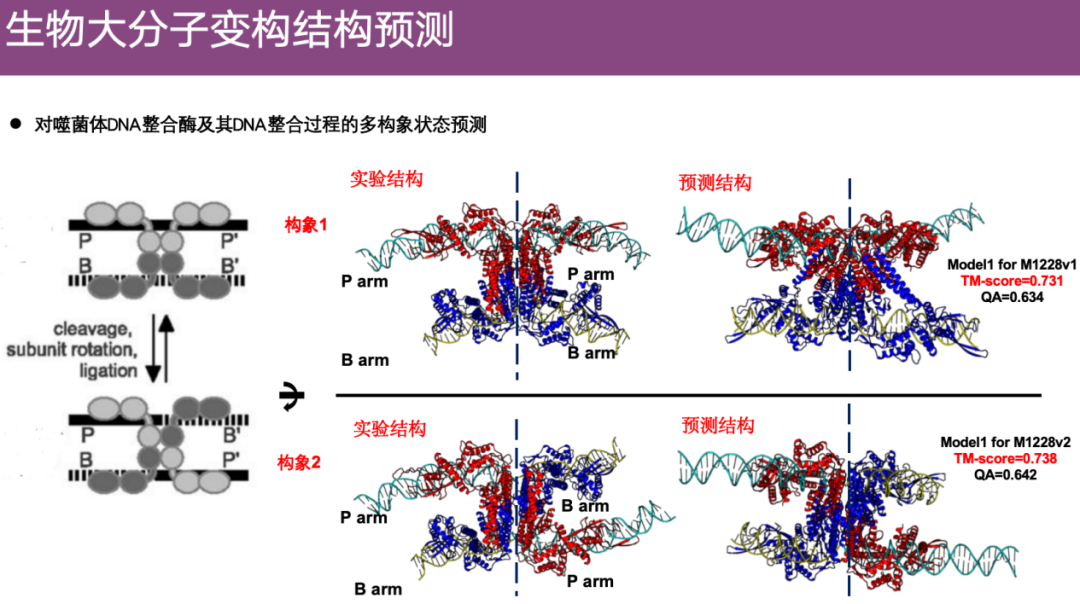
Il convient de mentionner que dans le concours CASP 16,Les candidats n’ont reçu que des informations sur la séquence et ne connaissaient pas le processus biologique spécifique ni les détails des changements conformationnels. Cependant, l’équipe du professeur Zheng Wei a réussi à restaurer l’ensemble du processus biologique grâce à la prédiction.Lors du résumé d'après-match, les juges ont également exprimé leur surprise.
Recrutement du groupe de recherche
Le professeur Zheng Wei de l'École de statistique et de science des données de l'Université de Nankai s'est depuis longtemps consacré à la recherche sur la prédiction de la structure, de la fonction et de l'interaction des macromolécules biologiques telles que les protéines. Il a dirigé le développement d'un certain nombre d'algorithmes de prédiction de structure de monomères protéiques, de complexes protéiques, d'acides nucléiques et de complexes, de complexes protéine-acide nucléique et d'algorithmes d'évaluation de structure avec une meilleure précision qu'AlphaFold2/3. Il a remporté le championnat dans de nombreuses compétitions du World Protein Structure Prediction Competition (CASP) (CASP13-16), dirigeant plus de 80 groupes de recherche universitaires/industriels à travers le monde.
L'équipe de bioinformatique de l'École de statistique et de science des données de l'Université de Nankai où il travaille recrute de nouveaux membres.Si vous êtes intéressé par la biologie structurale computationnelle, la bioinformatique ou la science des données, que vous soyez titulaire d'une maîtrise, d'un doctorat ou d'un postdoctorat, vous êtes le bienvenu pour rejoindre l'équipe du professeur Zheng Wei.
Les étudiants intéressés peuvent contacter le professeur Zheng Wei via les méthodes suivantes :
* Courriel : [email protected]
* WeChat : 18622152765









