Command Palette
Search for a command to run...
Le Taux De Réussite Peut Atteindre 100%. La Société De Développement De Médicaments Cellarity s'est Associée À NVIDIA Pour Optimiser Des Molécules Ciblées Basées Sur l'apprentissage Par Renforcement

Depuis l’Antiquité jusqu’à nos jours, l’humanité n’a jamais cessé de lutter contre les maladies. L’émergence d’un nouveau médicament pourrait sauver des milliers de vies et même prolonger la durée de vie humaine globale.
En repensant à l’histoire centenaire de la recherche et du développement de médicaments, on découvre de nombreuses histoires intéressantes. Par exemple, au début du XIXe siècle, l'assistante d'un pharmacien allemand, Zeltina, faisait tremper de l'opium dans de l'eau chaude, puis l'extrayait avec de l'eau ammoniacale pour séparer un tas de poudre blanche de l'opium. Il a donné cette poudre blanche au chien, et le chien s'est rapidement évanoui après l'avoir mangée.Il l'a donc nommé morphine, d'après le dieu grec des rêves, Morphée.La morphine est donc généralement considérée comme le premier ingrédient actif au monde isolé d’une plante et comme le point de départ de l’innovation pharmaceutique moderne.
Par la suite, les pharmaciens ont progressivement maîtrisé la technologie de synthèse des médicaments chimiques, et le pharmacien allemand Selmann a synthétisé l'acide acétylsalicylique, le prédécesseur de l'aspirine. Au début du XXe siècle,La demande des entreprises en nouveaux médicaments a stimulé le développement d’une technologie de criblage à haut débit, qui permet aux scientifiques de cribler et de tester un grand nombre de composés avec une plus grande efficacité. Au début du 21e siècle,Les chercheurs ont commencé à explorer des traitements médicamenteux plus précis et plus efficaces, parmi lesquels les médicaments ciblés sont devenus une direction de recherche en vogue.
Aujourd’hui, le développement rapide de la technologie de l’intelligence artificielle a apporté de nouvelles possibilités à la découverte de médicaments. L’IA peut aider les pharmaciens à valider plus rapidement les cibles médicamenteuses et à optimiser la conception de la structure des médicaments, et même à générer directement des molécules dotées de propriétés physico-chimiques ou d’activités biologiques spécifiques, accélérant ainsi considérablement la découverte de médicaments.
Dans ce contexte,Des chercheurs de la société des sciences de la vie Cellarity et NVIDIA ont proposé conjointement une nouvelle méthode d'optimisation moléculaire ciblée basée sur l'apprentissage par renforcement latent, MOLRL.L'approche combine un puissant modèle génératif pré-entraîné sur un grand ensemble de données chimiques avec un algorithme d'apprentissage par renforcement (RL) de pointe pour une optimisation continue de l'espace. En appliquant la méthode aux tâches liées à la découverte de médicaments, en utilisant des critères de référence communs et en la comparant aux méthodes de pointe, les chercheurs ont découvert que MOLRL présentait des performances supérieures ou compétitives dans une variété de tâches, en particulier dans la génération de molécules ciblées et l'optimisation multiparamétrique.
Les résultats associés ont été publiés sur ChemRxiv sous le titre « Génération moléculaire ciblée avec apprentissage par renforcement latent ».
Adresse du document :
Suivez le compte officiel et répondez « optimisation moléculaire ciblée » pour obtenir le PDF complet
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Sélection de la voie : modification directe des molécules ou fonctionnement dans l'espace latent
Le développement de médicaments est un processus très complexe : en plus de son activité biologique, un composé doit posséder de nombreuses autres propriétés pour être sélectionné comme candidat clinique. Les structures des composés identifiés comme ayant une activité thérapeutique, souvent appelés « composés candidats », ne sont pas gravées dans le marbre, mais sont modifiées au cours d’un long cycle itératif pour résoudre des problèmes tels qu’une solubilité et une activité insuffisantes.
Dans un processus itératif, les pharmaciens transforment généralement les molécules initiales pour concevoir des analogues en fonction de leur intuition ou par énumération à partir de bibliothèques basées sur des réactions. Cependant, étant donné la taille même de l’espace chimique, la conception devient extrêmement difficile, même pour une seule molécule, nécessitant une évaluation exhaustive de l’ensemble de l’espace chimique. Les méthodes informatiques de génération de molécules ciblées peuvent explorer efficacement l’espace chimique et recommander aux chimistes des structures jusqu’alors inexplorées.
Actuellement, les méthodes de génération et d’optimisation de molécules cibles peuvent être divisées en deux catégories :La première méthode consiste à opérer directement sur la structure moléculaire.identifier les modifications structurelles susceptibles d’améliorer les propriétés cibles ;La deuxième catégorie de méthodes opère dans l’espace latent du modèle génératif.Modifier la structure moléculaire indirectement par sa représentation latente.
La méthode 1 peut effectuer des modifications structurelles en insérant ou en supprimant des atomes ou des liaisons chimiques, et l’industrie a fait des progrès considérables.
Il a été rapporté qu'en novembre de l'année dernière, une équipe dirigée par le professeur Yoonsu Park de l'Institut coréen avancé des sciences et technologies (KAIST) a développé une technologie innovante d'édition d'atomes uniques. Cette technologie introduit des photocatalyseurs.L’édition d’un seul atome de molécules de médicaments a été réalisée avec succès à température et pression ambiantes.La technologie des « ciseaux moléculaires » développée par l'équipe peut couper et connecter avec précision des structures cycliques à cinq éléments, remplaçant les atomes d'oxygène par des atomes d'azote, modifiant les propriétés moléculaires et améliorant l'efficacité du médicament. Les résultats de recherche pertinents ont été publiés dans Science sous le titre « Conversion photocatalytique du furane en pyrrole ».

Cependant, il n’est pas facile de réaliser une intervention chirurgicale sur des molécules à volonté. D’une part, les modifications structurelles peuvent violer les règles de la chimie et ainsi conduire à des structures moléculaires invalides. D’autre part, comme les structures moléculaires sont intrinsèquement discrètes et que l’ajout ou la suppression de liaisons chimiques implique des opérations discrètes, cette discrétion conduira à des gradients discontinus dans le processus d’optimisation, ce qui rend difficile l’application efficace des méthodes basées sur les gradients.
Par rapport à la méthode 1,La deuxième approche transforme la tâche d’optimisation en un problème d’optimisation continue, exploite l’espace latent du modèle génératif et adopte des algorithmes d’optimisation d’espace continu tels que la descente de gradient.Néanmoins, la validité chimique reste un défi car il n’y a aucune garantie qu’un point dans l’espace latent corresponde à une molécule valide. Cependant, en utilisant de nouvelles architectures ainsi que des modifications de formation, les modèles génératifs ont fait des progrès significatifs dans l’amélioration de l’efficacité et de la continuité dans l’espace latent.
Dans le cadre de la recherche de Cellarity et NVIDIA, les chercheurs ont proposé MOLRL pour optimiser l'espace latent du modèle génératif pré-entraîné en utilisant la méthode d'optimisation de politique proximale (PPO).
MOLRL, une méthode d'optimisation moléculaire ciblée basée sur l'apprentissage par renforcement latent
Comment fonctionne le cadre MOLRL ?
Le cadre MOLRL se compose de deux parties : un modèle génératif d'espace latent et un agent d'apprentissage par renforcement (RL).
Le modèle génératif est un modèle encodeur-décodeur pré-entraîné dont l'espace latent code l'espace chimique sur lequel opère l'agent RL. L'agent RL est formé à l'aide de la méthode PPO,Naviguer dans l'espace latent; La fonction de récompense fournit un retour d'information à l'agent,Aidez-les à apprendre à naviguer dans l’espace,Identifier les molécules possédant les propriétés souhaitées.
Comme indiqué ci-dessous : la représentation latente « z » de la molécule d’entrée est perturbée par l’action « a » extraite de la sortie du réseau de politiques. Le vecteur latent perturbé « z′ » est décodé en molécules et noté par une fonction de récompense. L'état « z », l'action « a » et la récompense « R » sont collectés et utilisés pour mettre à jour le réseau de politiques.
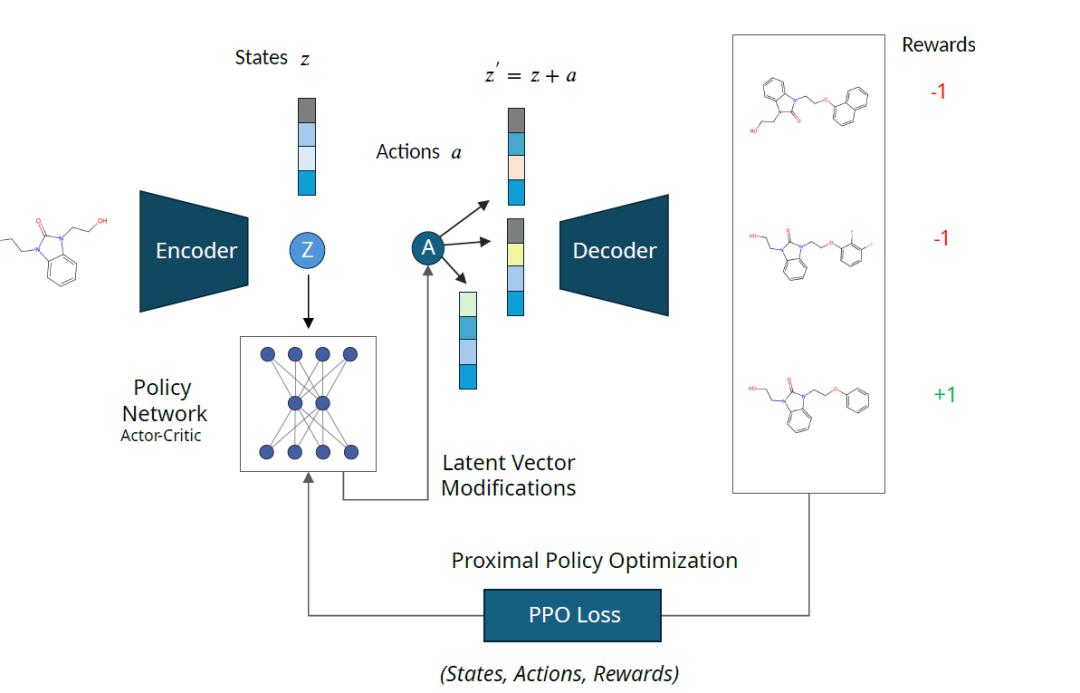
Le cadre est indépendant de l'architecture de l'encodeur et du décodeur, cependant, les caractéristiques de l'espace latent affecteront grandement les performances d'optimisation. Par conséquent, les chercheurs ont évalué les performances de MOLRL sur deux architectures d'encodeur-décodeur différentes, à savoir l'autoencodeur variationnel (VAE) et l'autoencodeur formé sur la base de l'apprentissage automatique d'informations mutuelles (MolMIM).
Un agent d’apprentissage par renforcement (RL) est chargé de naviguer dans l’espace latent pour identifier les molécules possédant les propriétés moléculaires souhaitées. Les chercheurs ont utilisé PPO, ou Proximal Policy Optimization, pour former l'agent RL.L'algorithme PPO guide l'agent pour trouver le chemin optimal dans l'espace latent en optimisant la politique pour maximiser la récompense cumulative à long terme.La fonction de récompense est au cœur du cadre MOLRL, qui fournit un retour d'information à l'agent en fonction des propriétés cibles de la molécule (telles que la similarité avec le médicament, l'accessibilité synthétique, la liaison à la cible, etc.).
Comment fonctionne le framework MOLRL ?
Pour évaluer les performances du cadre MOLRL, les chercheurs ont conçu une tâche d’optimisation multi-objectifs et l’ont comparée aux méthodes d’optimisation de pointe actuelles.
Plus précisément, les chercheurs ont appliqué MOLRL pour générer des molécules biologiquement actives ciblant deux cibles tout en optimisant à la fois la similarité médicamenteuse (QED) et l'accessibilité synthétique (SA). Les cibles biologiques choisies étaient deux kinases associées à la maladie d'Alzheimer - GSK3β et JNK3. Sur la base de la stratégie d'évaluation de Jin et al., les chercheurs ont enregistré les 5 000 molécules ayant les valeurs de récompense les plus élevées générées au cours du processus d'optimisation et ont calculé les trois indicateurs suivants : taux de réussite ; nouveauté; et la diversité.
Le tableau suivant montre les performances de MOLRL formé dans l'espace latent VAE-CYC et de MOLRL formé dans l'espace MolMIM, ainsi que la comparaison des performances des méthodes d'optimisation moléculaire de pointe actuelles rapportées dans la littérature.
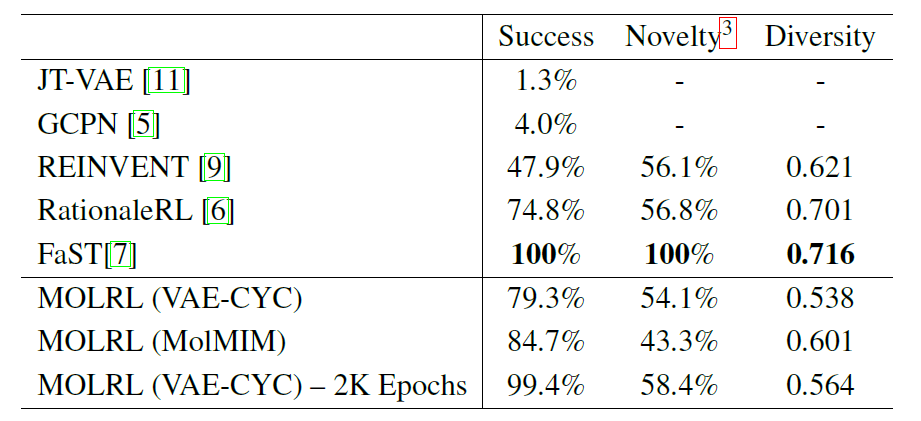
Comme le montre le tableau, FaST construit des graphiques moléculaires en combinant des fragments moléculaires à l’aide de l’apprentissage par renforcement (RL).Il montre un taux de réussite plus élevé parmi toutes les méthodes comparées. FaST et RationaleRL présentent des avantages en termes de diversité et de nouveauté, et les deux méthodes exploitent les connaissances antérieures. REINVENT et MOLRL partent tous deux de molécules aléatoires qui peuvent être éloignées de la plage d'entraînement du classificateur ML.MOLRL atteint toujours une nouveauté comparable à RationaleRL et obtient le taux de réussite le plus élevé.
Utiliser les connaissances préalables comme point de départ peut présenter certains avantages, mais cela peut également limiter la nouveauté et la capacité de l’algorithme à découvrir de nouveaux squelettes. De plus, l’applicabilité de telles méthodes est limitée lorsqu’aucune connaissance préalable n’est disponible, comme lors de l’étude de cibles inexplorées.
Outre les tâches d’optimisation multi-objectifs, une approche courante dans la découverte de médicaments consiste à identifier un échafaudage chimique connu pour se lier à une certaine cible ou classe de cibles et à l’utiliser comme point de départ pour la conception et l’optimisation chimiques. Par conséquent, l’article vérifie davantage la capacité du MOLRL à optimiser les propriétés multi-objectifs tout en préservant le squelette moléculaire spécifié. Comme le montre le tableau suivant,Lors de l'optimisation de molécules contenant un squelette aminopyrimidine, MOLRL a obtenu un taux de réussite de 100%.
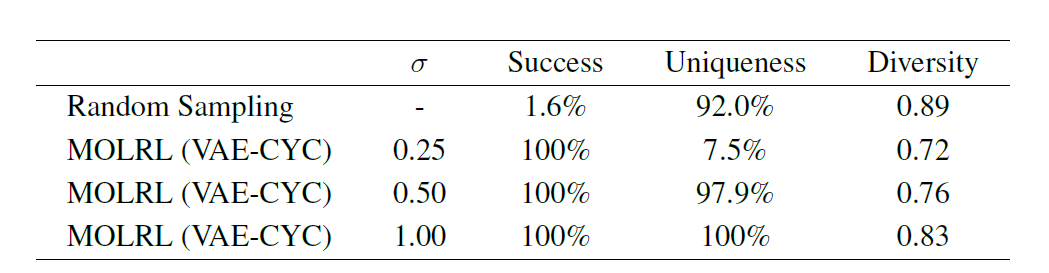
En résumé, MOLRL montre des performances supérieures ou compétitives sur une variété de tâches par rapport aux méthodes existantes.Notamment dans la génération de molécules ciblées et l'optimisation multiparamétrique.
L’IA est une étape clé pour améliorer l’efficacité de la découverte de médicaments
Combien de ressources faut-il pour développer un nouveau médicament ? L'industrie pharmaceutique a une célèbre « règle du double dix », selon laquelle il faut 10 ans et 1 milliard de dollars pour qu'un nouveau médicament soit découvert et mis sur le marché. Selon le dernier rapport publié par Deloitte, si l'on prend en compte le coût des essais cliniques ratés, le coût moyen pour les plus grandes sociétés pharmaceutiques mondiales de mettre avec succès un nouveau médicament sur le marché est deIl est passé de 1,188 milliard de dollars en 2010 à 2,284 milliards de dollars en 2022.
Une étape clé dans la découverte de médicaments consiste à trouver un lot de molécules candidates pour une étude informatique ou une synthèse et une caractérisation, ce qui est une tâche difficile car l’espace chimique des molécules potentielles est énorme et nécessite des coûts d’essais et d’erreurs extrêmement élevés. Aujourd’hui, l’intelligence artificielle et l’apprentissage automatique peuvent améliorer efficacement l’efficacité de cette étape.
31 octobre 2023Les instituts Novartis pour la recherche biomédicale et le centre de recherche Microsoft pour l'intelligence scientifique ont collaboré pourUn article de recherche intitulé « Extraire l’intuition de la chimie médicinale via l’apprentissage automatique des préférences » a été publié dans Nature Communications.
Les chercheurs ont demandé à 35 chimistes médicinaux de choisir leur molécule préférée parmi 5 000 paires de molécules, ont utilisé leurs réponses pour former un jeu de classement afin de former un modèle d'apprentissage automatique, puis ont demandé au modèle de noter les molécules. Ce score n’est en grande partie pas affecté par d’autres propriétés qui étaient auparavant caractéristiques du domaine, car il provient d’années de connaissances accumulées au sein de l’industrie.
Le modèle peut reproduire partiellement les connaissances collectives accumulées par les chimistes professionnels dans leur travail, souvent appelées « intuition chimique », rendant le développement futur de médicaments plus efficace.
En mars 2024, Insilico Medicine, une société pharmaceutique leader dans le domaine de l'IA, a publié un article de recherche scientifique dans Nature Biotechnology, détaillant l'utilisation d'une plate-forme d'intelligence artificielle pour découvrir la nouvelle cible TNIK pour le traitement de la FPI, et le processus ultérieur d'utilisation d'une plate-forme de chimie générative pour concevoir la molécule ISM001-055.
ISM001-055 est un inhibiteur de petite molécule, une première mondiale.Ciblage de TNIK (kinase interagissant avec Traf2/NCK) pour le traitement de la fibrose pulmonaire idiopathique (FPI). Insilicon Valley Silicon a déclaré que l'IA générative peut considérablement améliorer l'efficacité de la R&D, réduire les coûts de la R&D et augmenter le taux de réussite de la R&D dans les premières étapes de la R&D. Prenons l’exemple des molécules contre la fibrose pulmonaire idiopathique, depuis la découverte précoce de cibles jusqu’à l’identification de composés candidats précliniques,Il n’a fallu que 18 mois et 2,6 millions de dollars ont été investis dans la recherche et le développement.
Un rapport de recherche de Fortune Business Insights montre que la taille du marché mondial de l'intelligence artificielle dans la découverte de médicaments est de 3 milliards de dollars américains en 2022 et devrait passer de 3,54 milliards de dollars américains en 2023 à 7,94 milliards de dollars américains en 2030, avec un taux de croissance annuel composé de 12,21 %. À l’avenir, la technologie de l’IA a un grand potentiel pour entraîner des changements dans l’industrie pharmaceutique.
Références :
1.https://mp.weixin.qq.com/s/OL7TJQcUE-ubhUDyc7GBzQ
2.https://www.thepaper.cn/newsDetail_forward_29097303
3.https://news.bioon.com/article/6127e7234091.html
4.https://bydrug.pharmcube.com/news/detail/49720140c1e9d57ac3c7cfe20ef7f8be
5.https://mp.weixin.qq.com/s/UGAXWMhPlSg2hFnI5ghr1w









