Command Palette
Search for a command to run...
Simulateur De Dynamique Moléculaire Facile À Utiliser LAMMPS : Contrôle De Température Npt Pour Estimer Le Point De Fusion Du FCC Cu

Depuis sa sortie open source en 2004, LAMMPS est largement utilisé dans le domaine de la modélisation des matériaux. Son nom complet est Large-scale Atomic/Molecular Massively Parallel Simulator, qui a été développé par Sandia National Laboratories aux États-Unis.
LAMMPS peut être utilisé pour modéliser une variété de matériaux, y compris les matériaux à l'état solide (métaux, semi-conducteurs), les biomolécules, les polymères, etc. Il peut fournir une variété de modèles d'interaction de particules pour différents matériaux.
Plus important encore, LAMMPS peut être exécuté sur un seul processeur ou en parallèle en utilisant des techniques de passage de messages et de décomposition spatiale du domaine de simulation. Le code est conçu pour être facile à modifier ou à étendre avec de nouvelles fonctionnalités, et de nombreux modèles ont des versions qui offrent des performances accélérées sur les processeurs, les GPU et les Intel Xeon Phis.
En 2022, des centaines de personnes ont contribué à de nouvelles fonctionnalités de LAMMPS, et son nombre de lignes de code est passé de 50 000 en 2004 à 1 million en 2022.
Pour permettre à chacun d'expérimenter plus facilement ce logiciel classique de simulation de dynamique moléculaire, la section tutoriel du site Web officiel d'HyperAI a désormais lancé le « Tutoriel de démarrage LAMMPS : Estimation du contrôle de la température npt du point de fusion du Cu FCC », qui peut être exécuté à l'aide de la version CPU de LAMMPS.
Adresse du tutoriel:
Exemple d'effet :

Après avoir terminé ce tutoriel, vous serez capable de :
* Comprendre le processus de fonctionnement du contrôle de la température npt
* Utilisez les commandes dump et fix pour prétraiter les données
Essai de démonstration
Démarrer le conteneur
1. Connectez-vous à hyper.ai, accédez à la page Tutoriels, sélectionnez Tutoriel de démarrage LAMMPS : Estimation du point de fusion du FCC Cu à l'aide du contrôle de température NPT, puis cliquez sur Exécuter ce tutoriel en ligne.
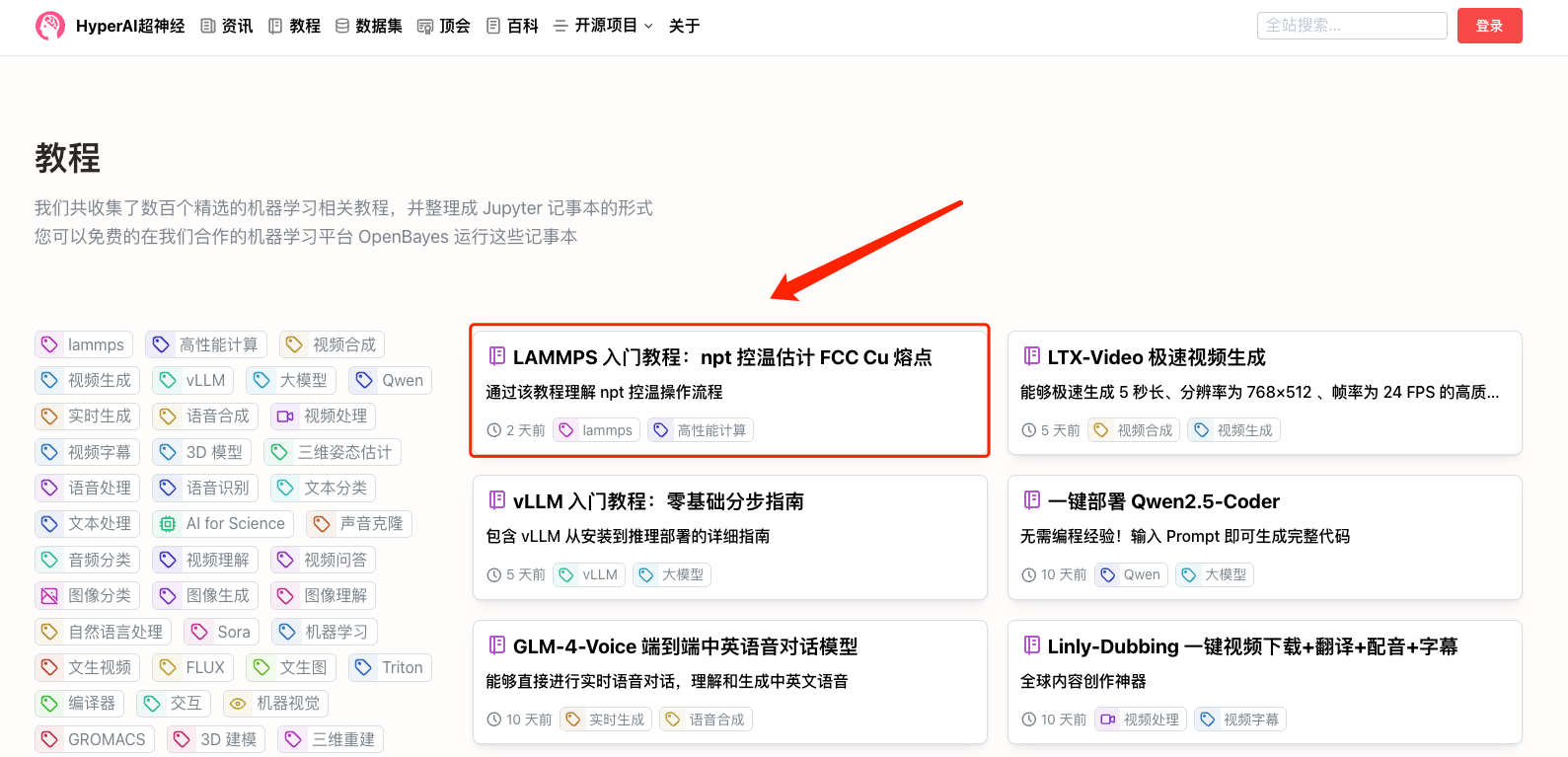
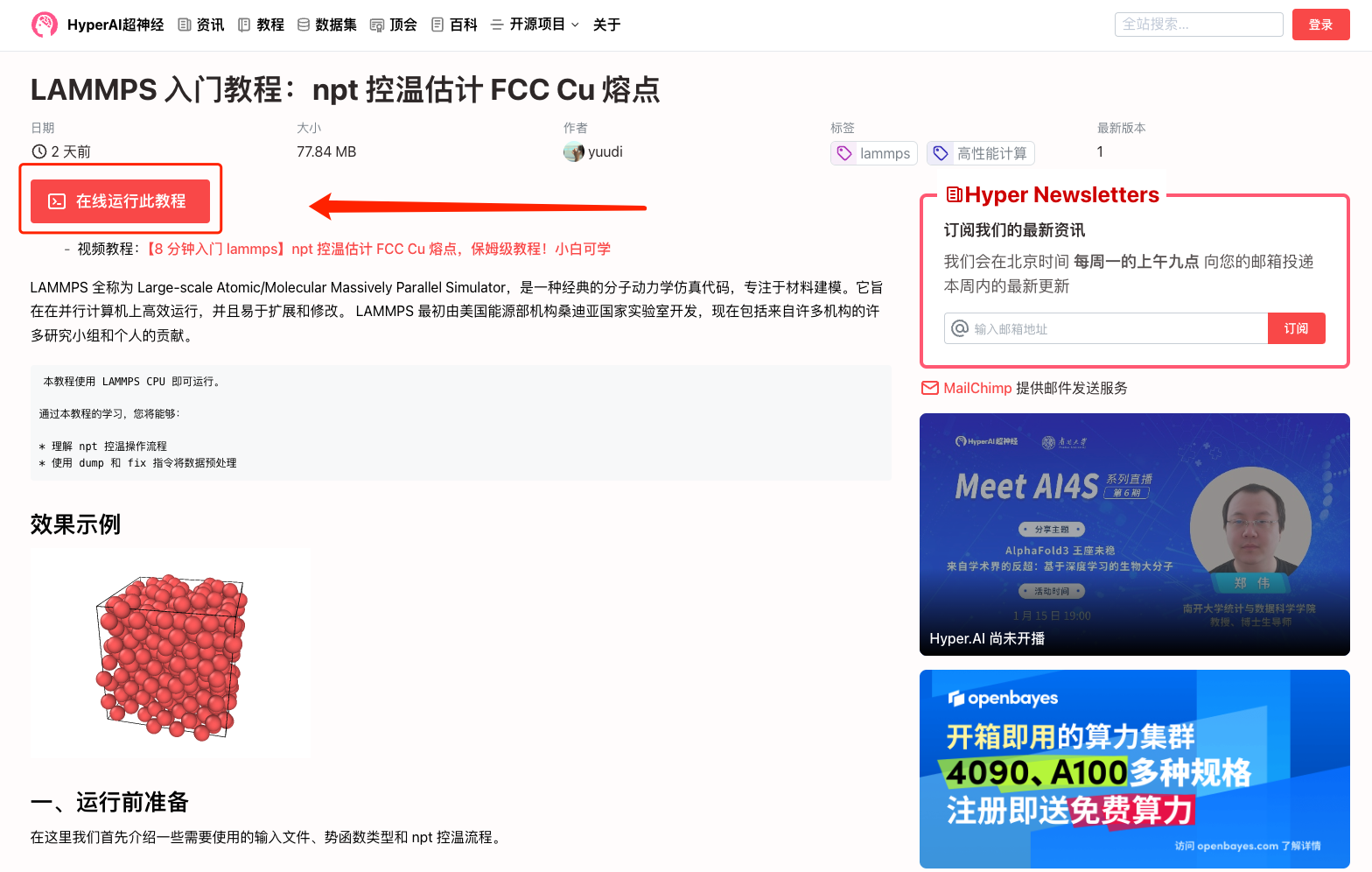
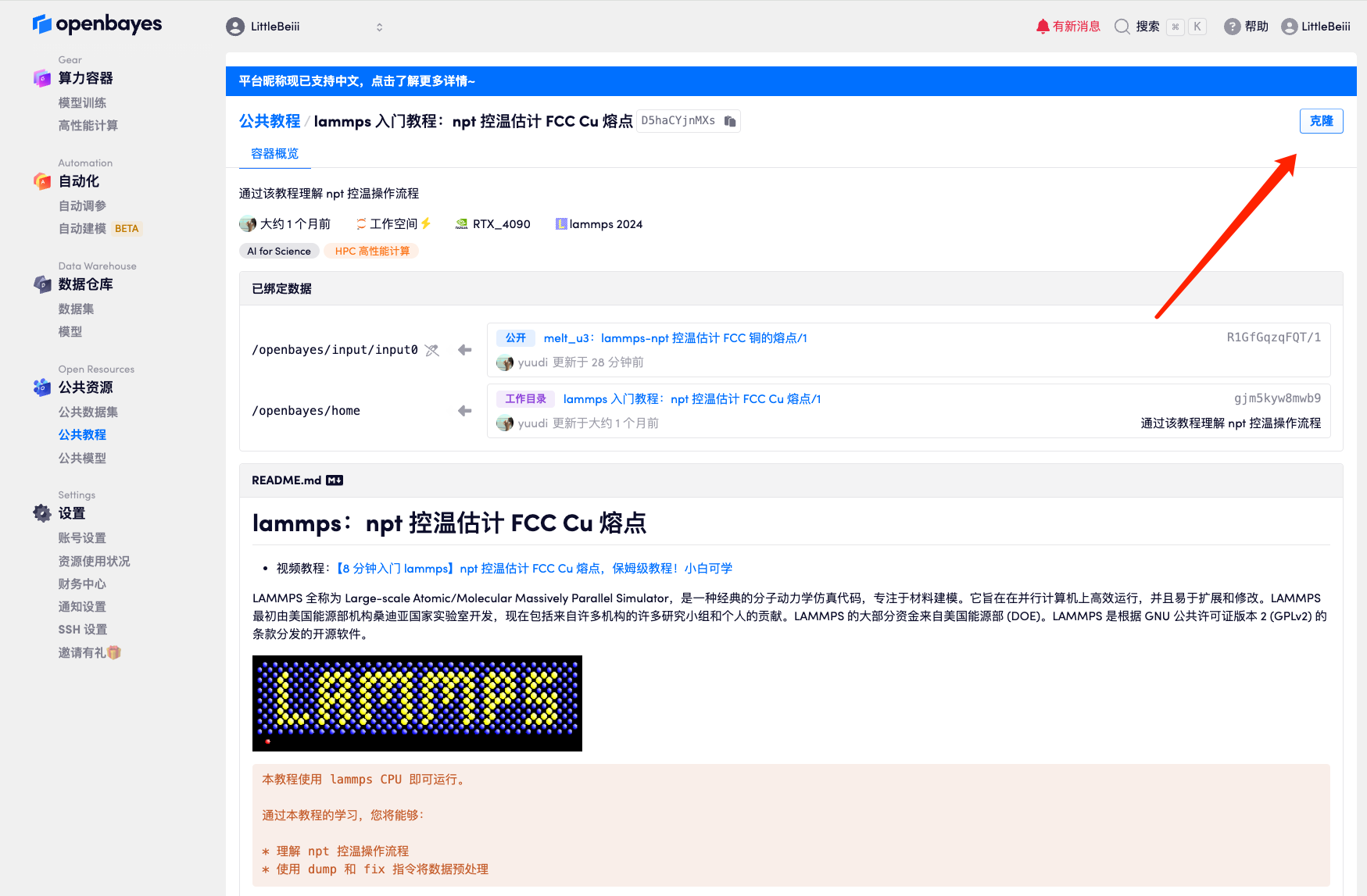
2. Une fois la page affichée, cliquez sur « Cloner » dans le coin supérieur droit pour cloner le didacticiel dans votre propre conteneur.
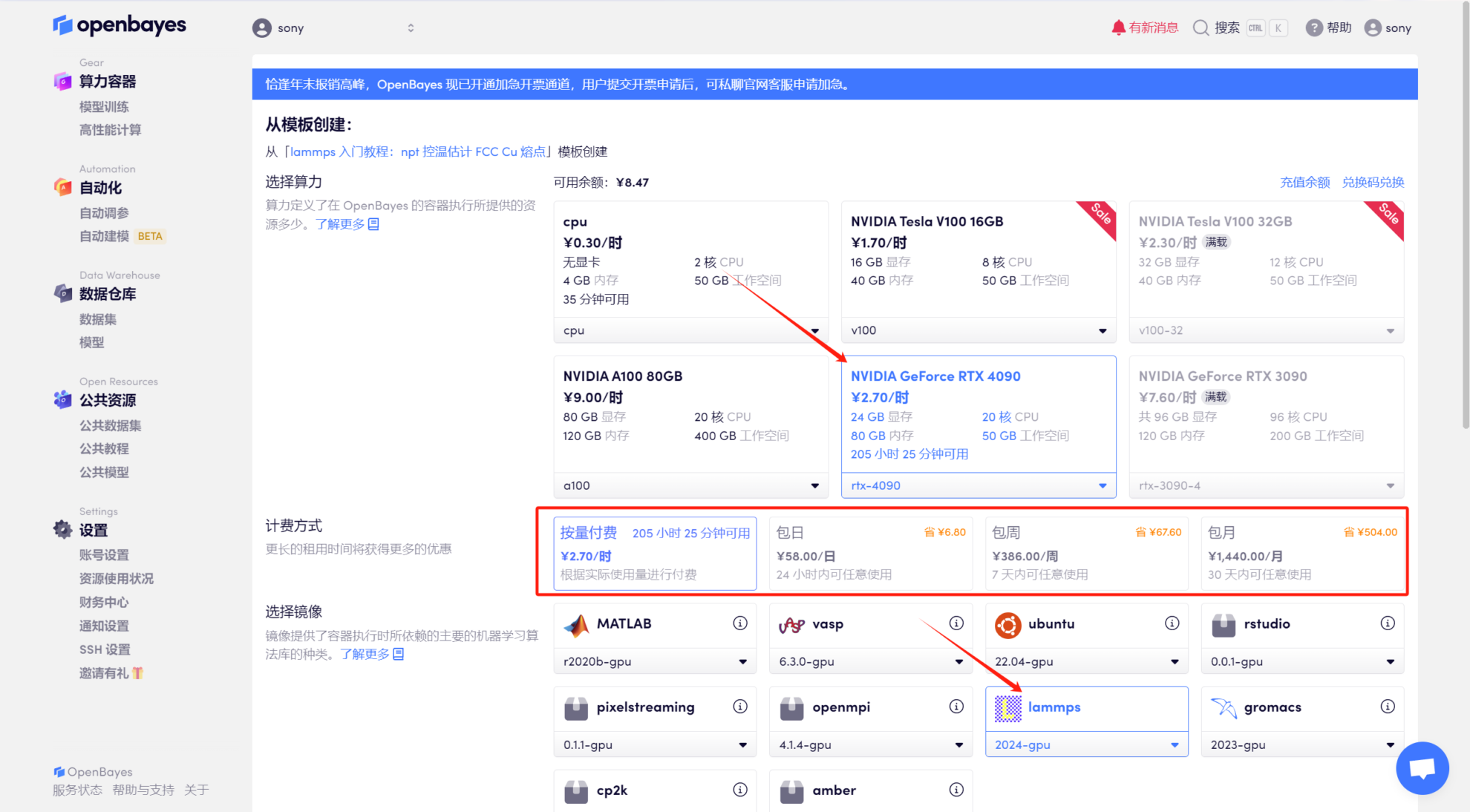
3. Sélectionnez la puissance de calcul « NVIDIA RTX 4090 », sélectionnez « Pay as you go » ou « Daily/Weekly/Monthly Package » selon vos besoins, sélectionnez « lampps » comme image, et enfin cliquez sur « Continuer ».
Les nouveaux utilisateurs peuvent s'inscrire en utilisant le lien d'invitation ci-dessous pour obtenir 4 heures de RTX 4090 + 5 heures de temps CPU gratuit !
Lien d'invitation exclusif HyperAI (copier et ouvrir dans le navigateur) :
https://openbayes.com/console/signup?r=Ada0322_QZy7
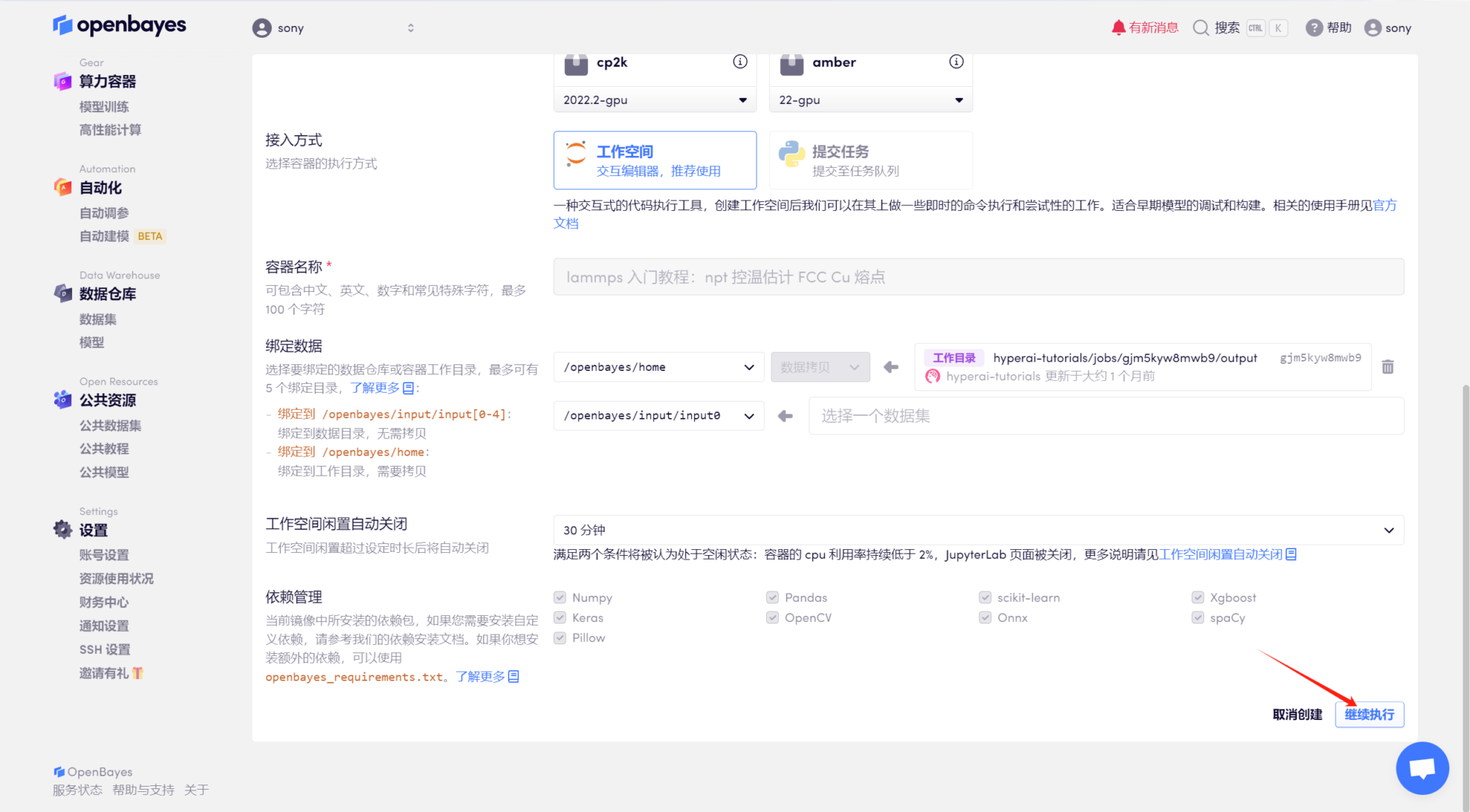
4. Attendez que le modèle alloue des ressources et que le statut passe à « En cours d'exécution », puis cliquez sur « Ouvrir l'espace de travail ».
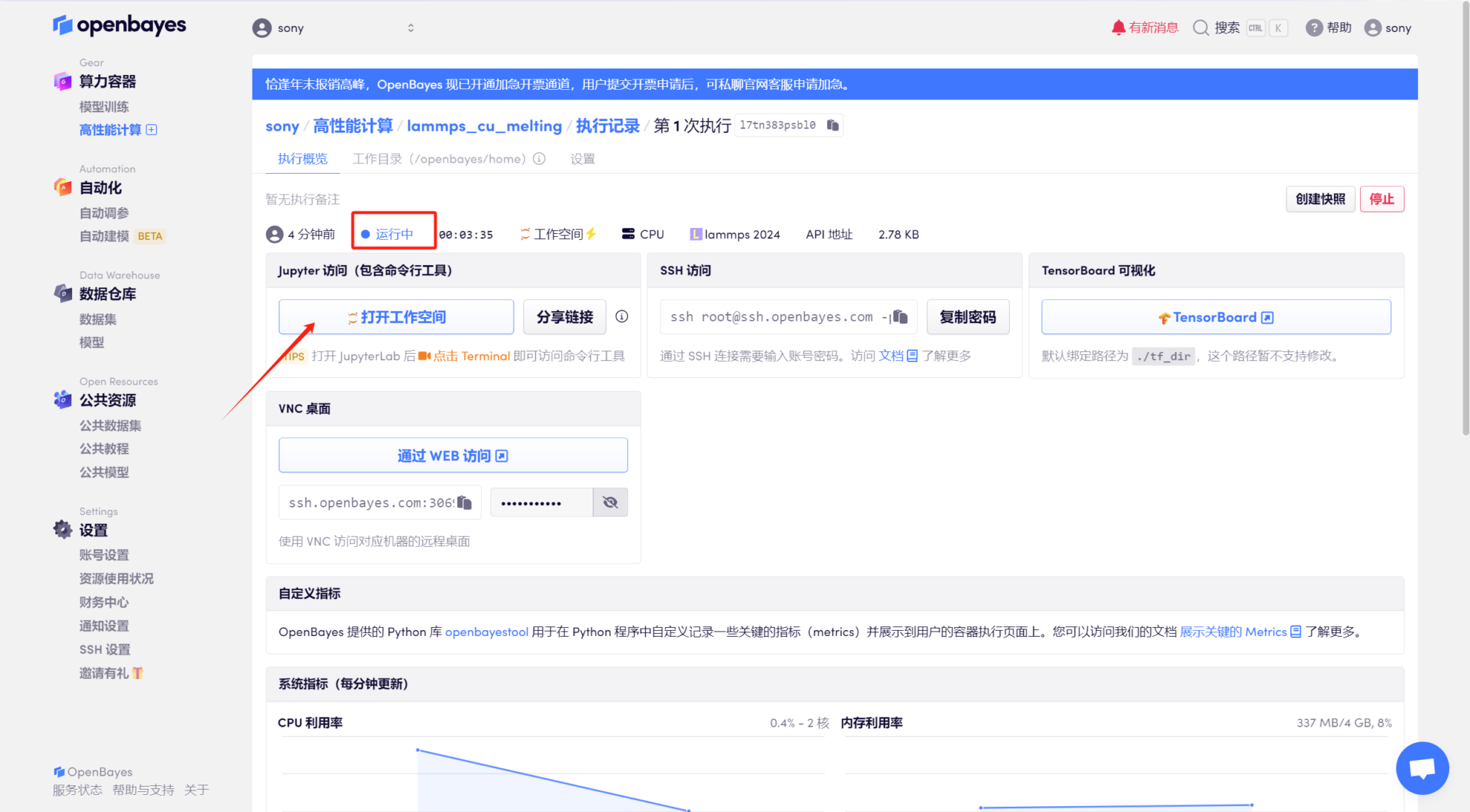
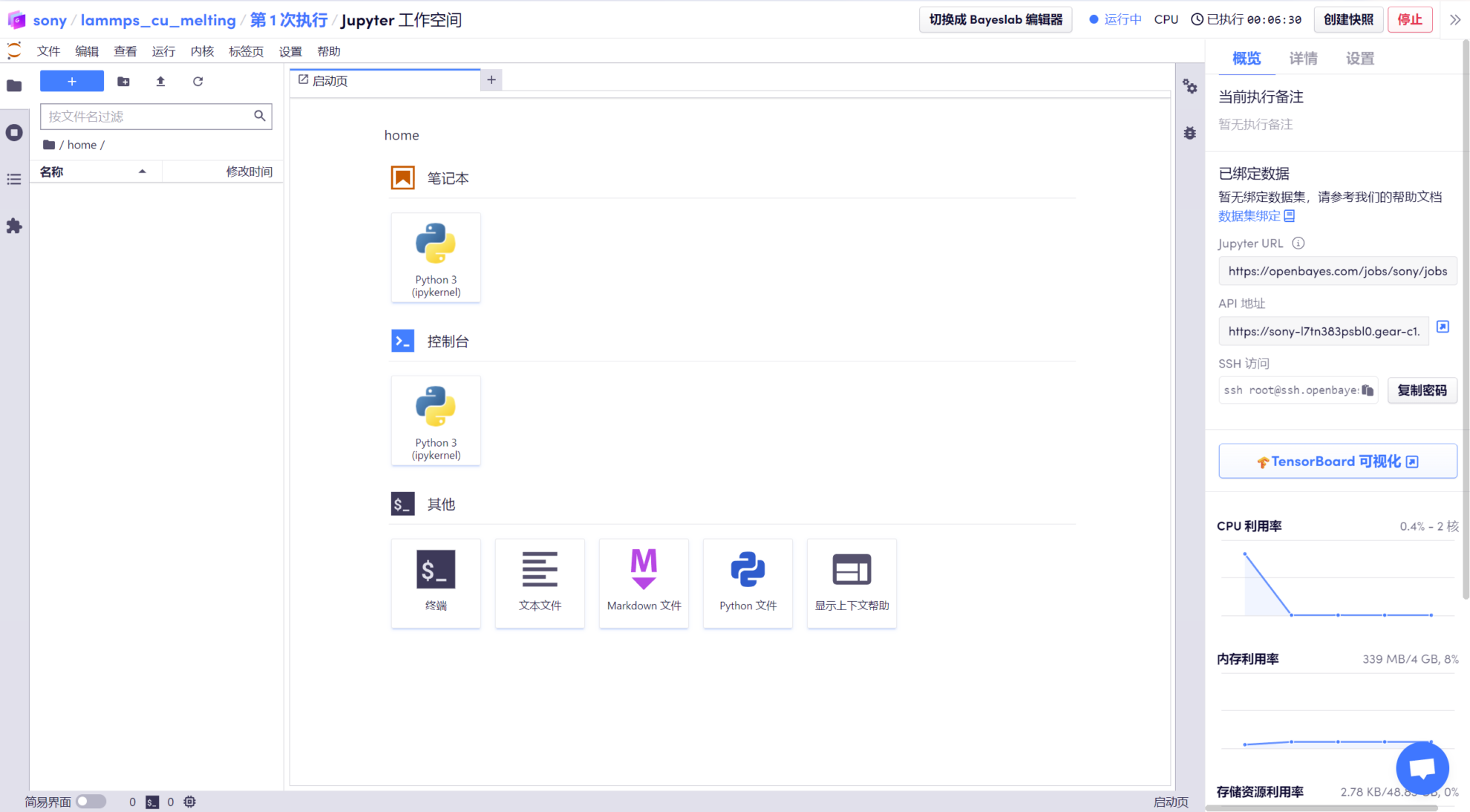
Étapes de course
1. Après être entré dans l'« Espace de travail », vous pouvez voir le package compressé « melt_u3.zip » préparé, qui contient les commandes pertinentes qui ont été saisies, telles que la définition du système, la lecture de la structure du cuivre, l'utilisation de la fonction de potentiel de brassage du cuivre, etc.
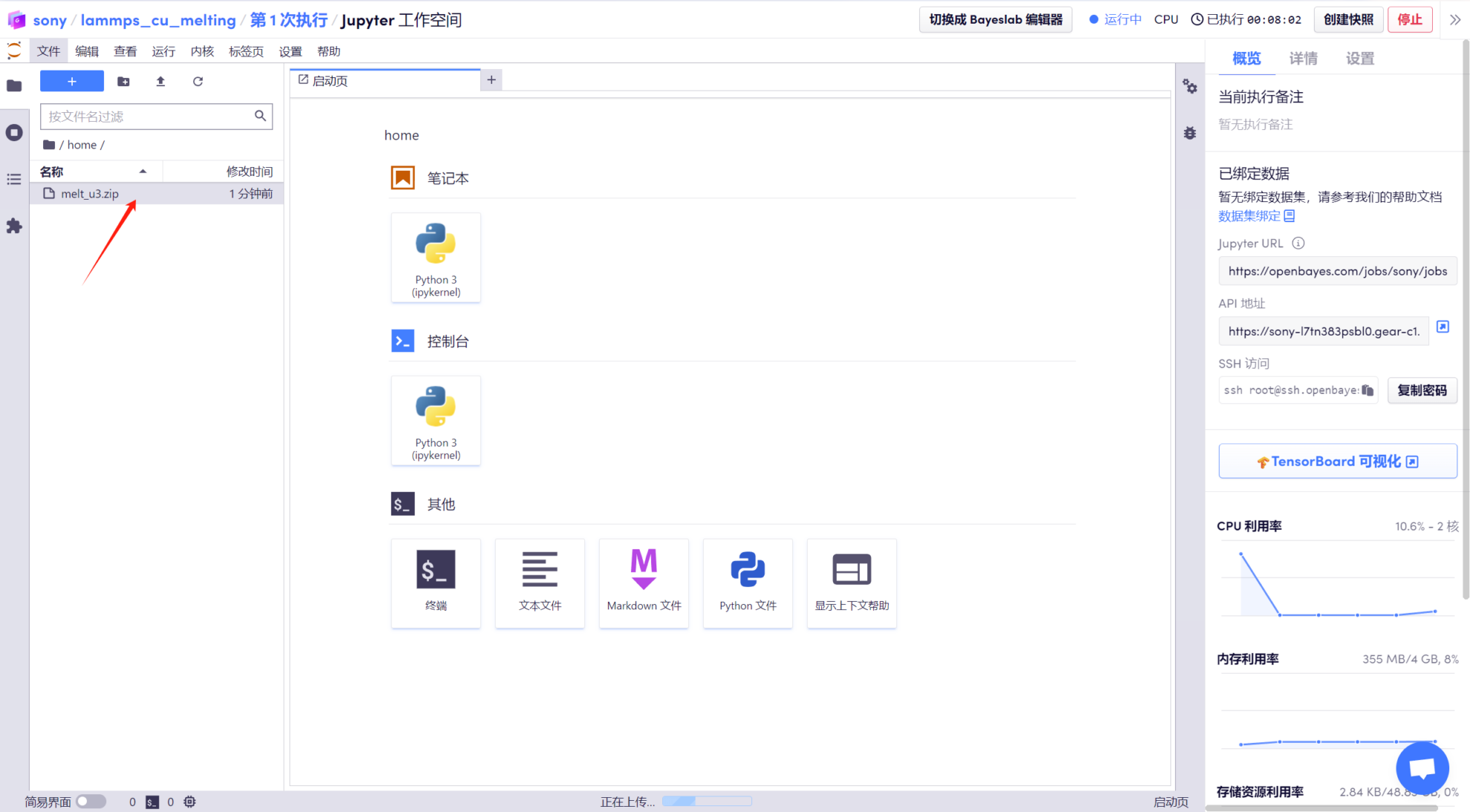
2. Ouvrez le « Terminal », entrez « cd melt_u3 » pour accéder au répertoire décompressé et utilisez la commande « ls » pour afficher les fichiers.
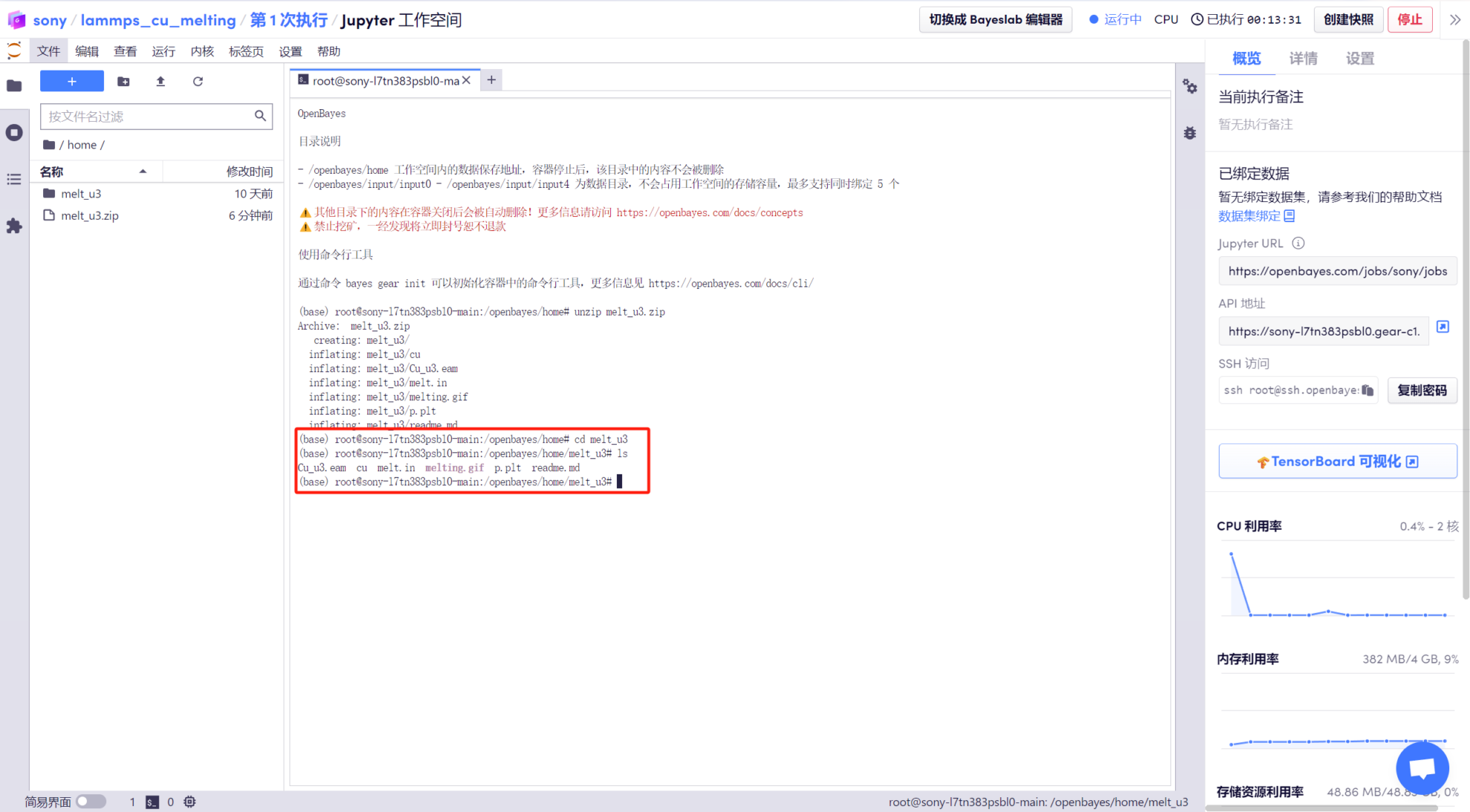
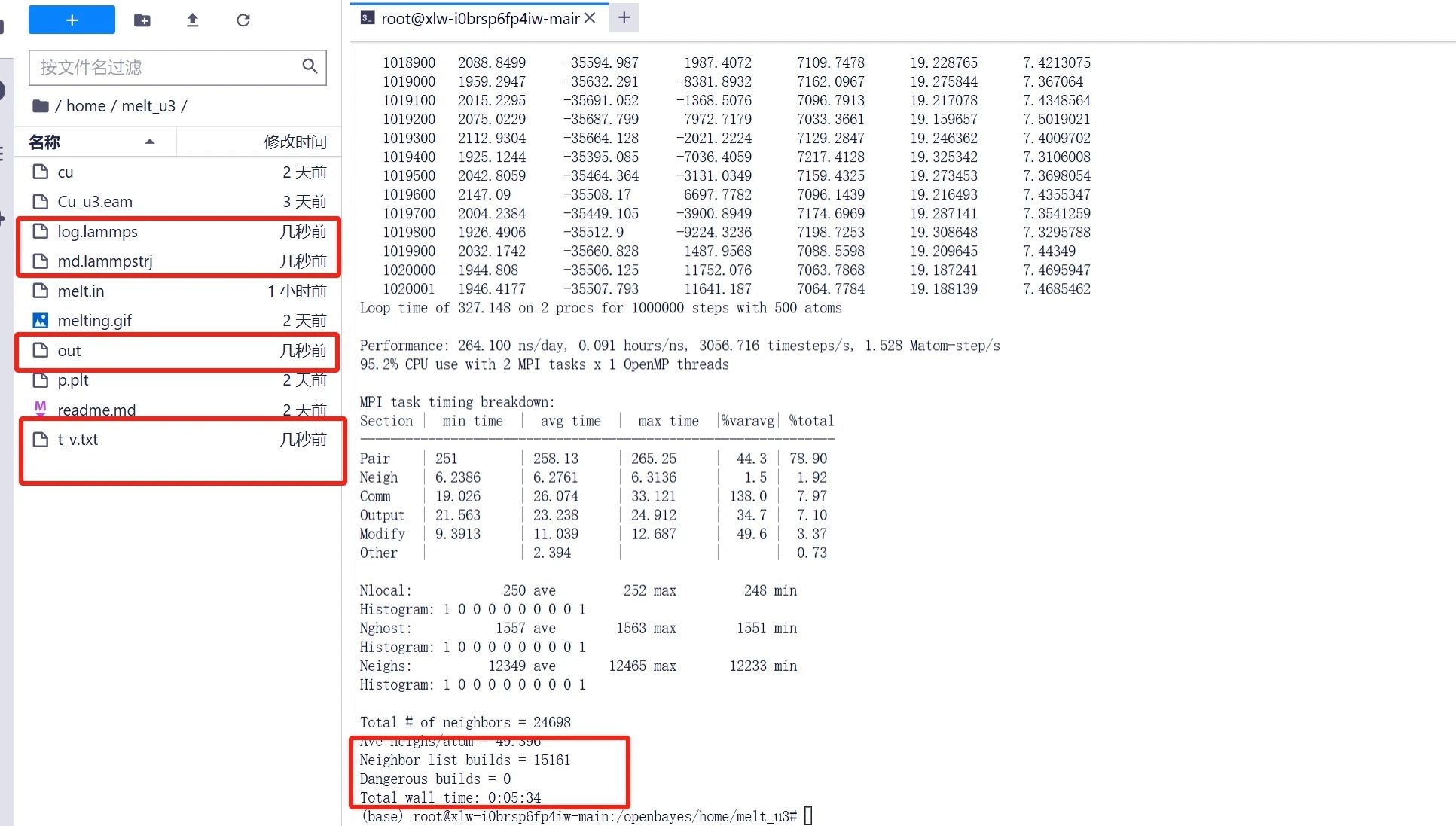
3. Entrez « mpirun -np 2 lmp < melt.in | tee out » pour exécuter lampps. L'ensemble du processus prend environ 5 à 10 minutes.
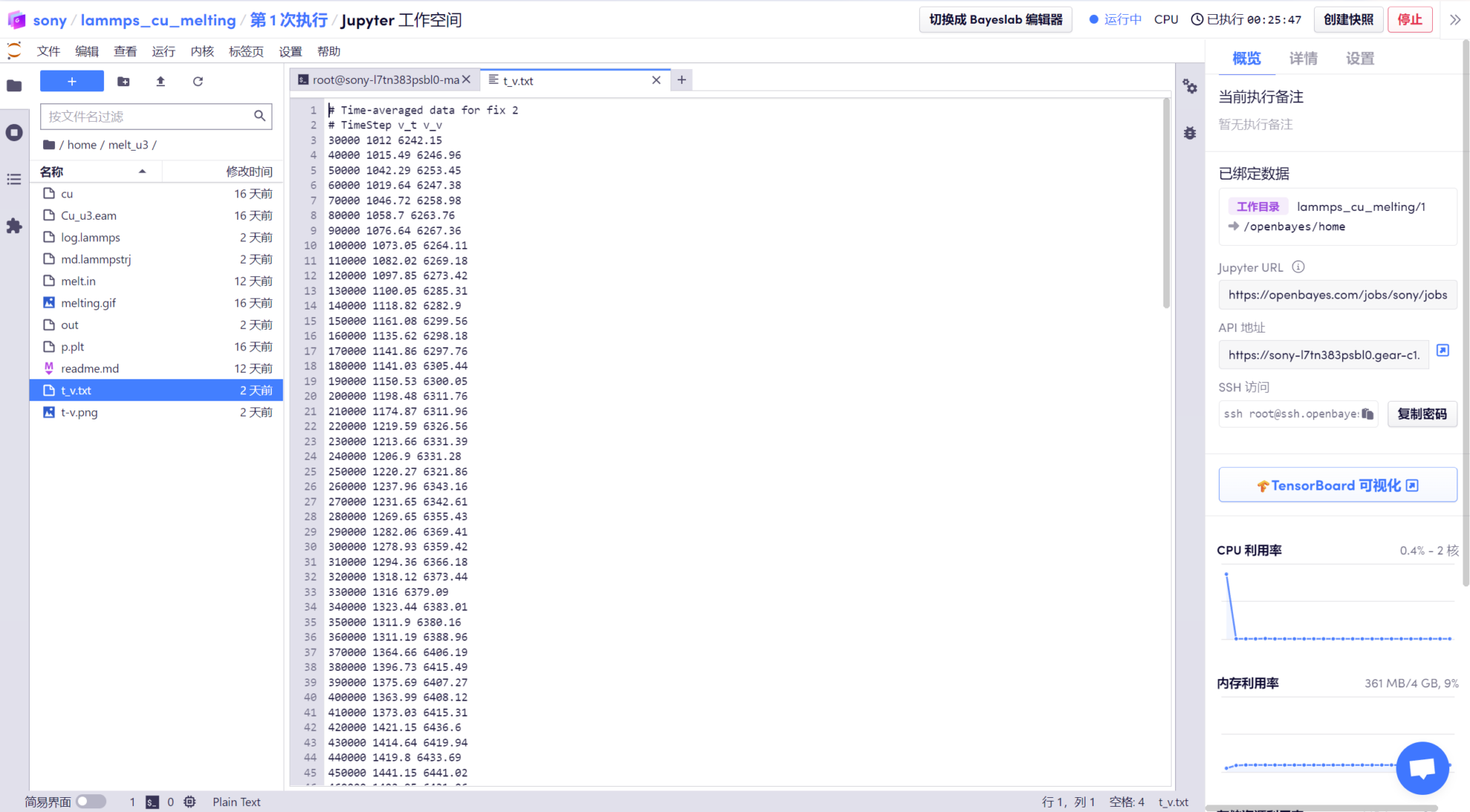
Une fois l'exécution terminée, vous pouvez obtenir des fichiers de sortie tels que « t_v.txt » dans le dossier. Les données de température et de volume sont saisies dans le fichier « t_v.txt ». Plus tard, nous devrons utiliser des outils de dessin pour visualiser les données.
Informatique
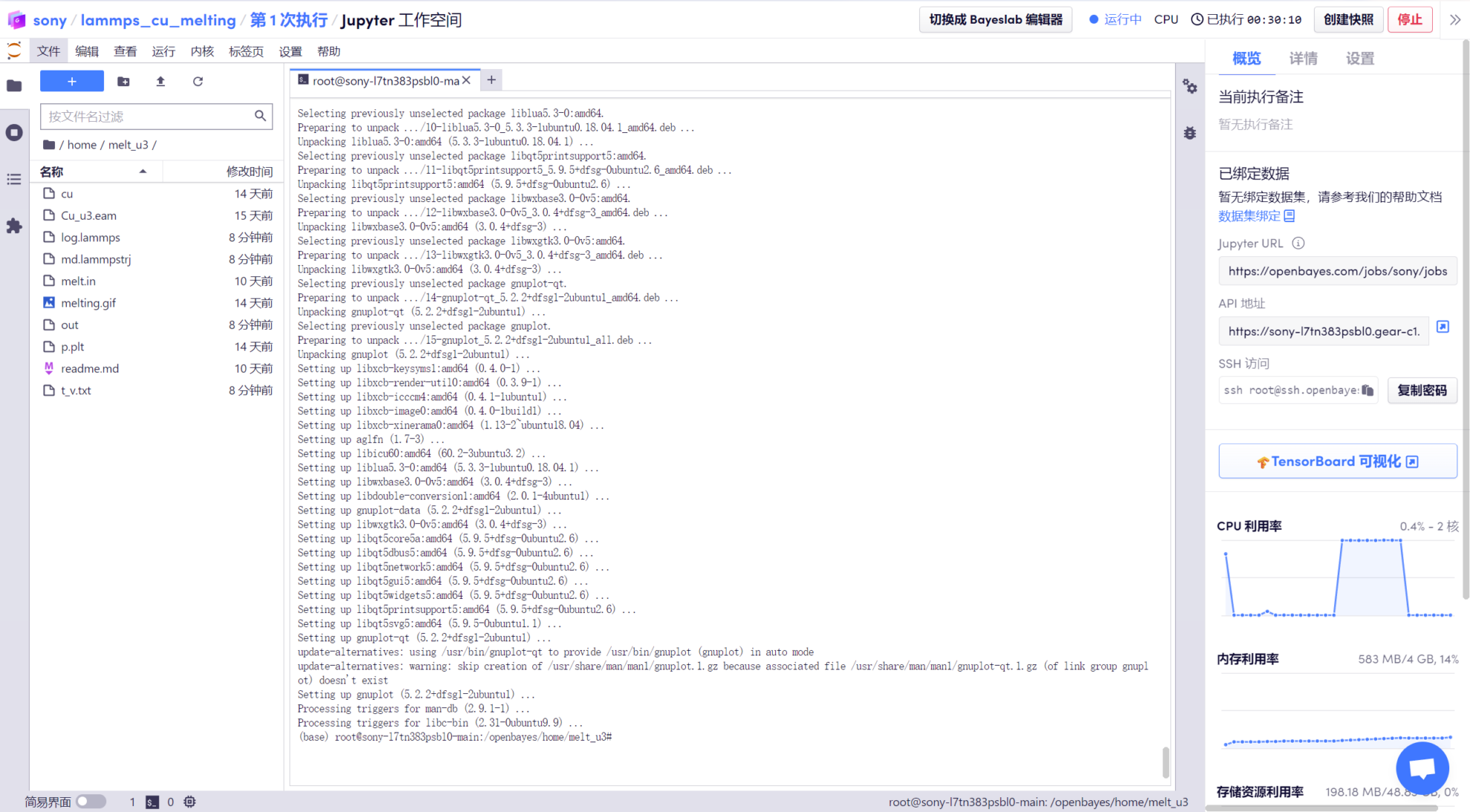
1. Après avoir attendu que le modèle s'exécute, entrez « apt-get update –fix-missing » pour mettre à jour la source apt. Après la mise à jour, entrez « apt install gnuplot » pour installer gnuplot (outil de dessin), puis entrez « y » et appuyez sur Entrée pour confirmer.
2. Utilisez l’outil de dessin nouvellement installé pour visualiser les données.
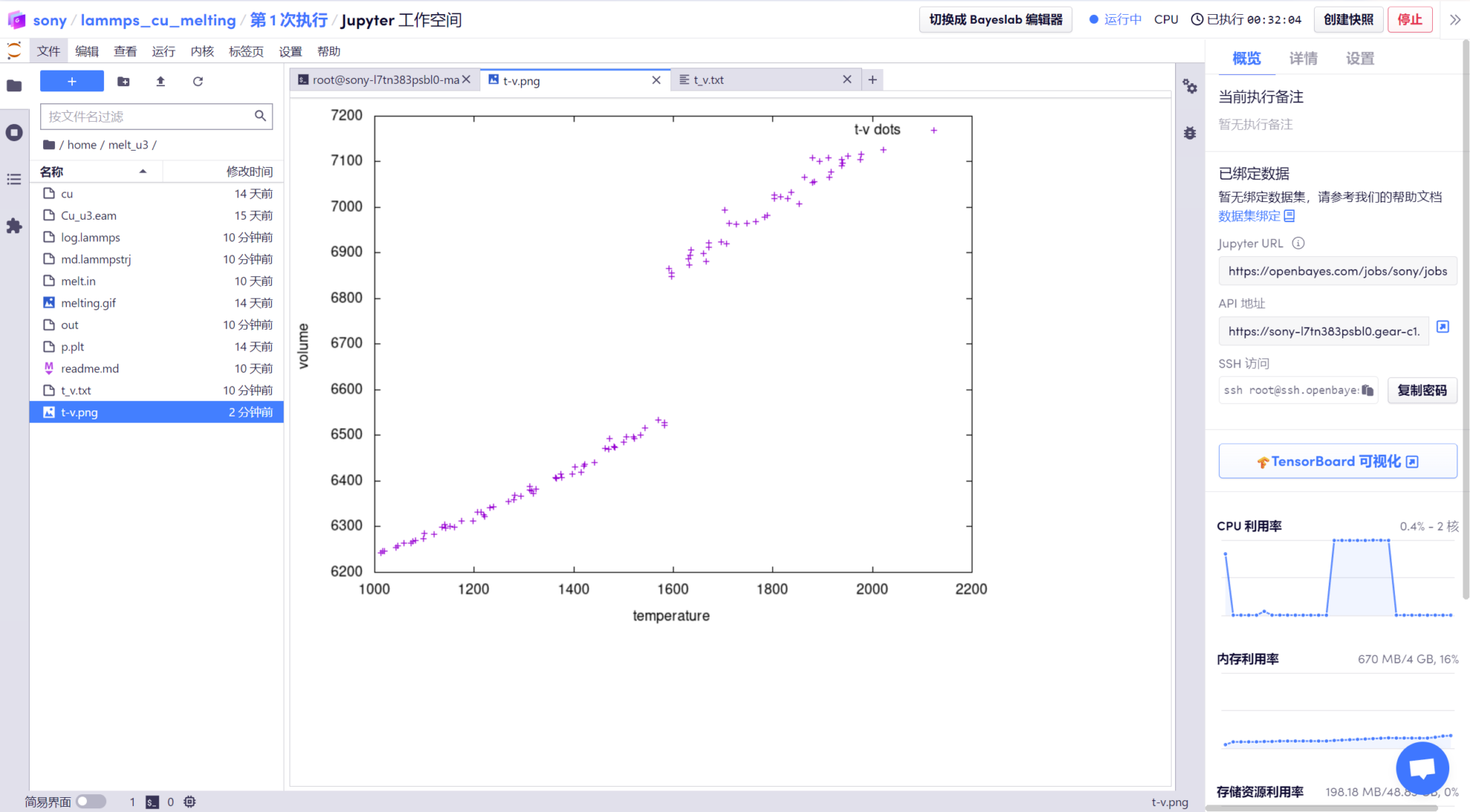
Le script d'exécution a été écrit. Exécutez simplement la commande « gnuplot p.plt » pour obtenir le graphique tv, qui est le graphique de la courbe d'étape température-volume. Vous pouvez voir que le point d'étape, c'est-à-dire le point de fusion, est de 1600K.
3. Téléchargez ensuite son fichier de trajectoire atomique « md.lammpstrj » sur votre ordinateur local.
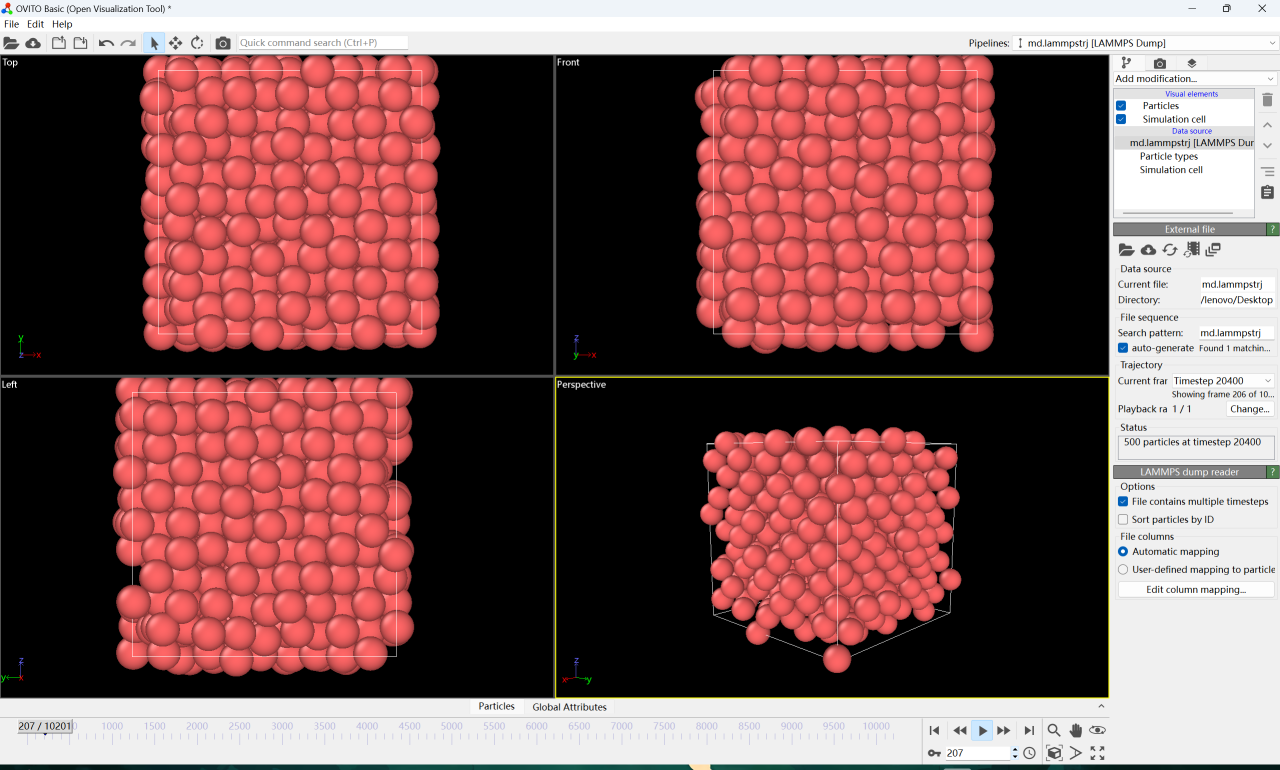
4. Ouvrez le fichier téléchargé dans OVITO et cliquez sur lecture pour voir la trajectoire de chaque atome de cuivre pendant le processus de dissolution du cuivre.
Nous avons créé un « Groupe d'échange de tutoriels de diffusion stable ». Bienvenue aux amis pour rejoindre le groupe pour discuter de divers problèmes techniques et partager les résultats des applications~
Scannez le code QR ci-dessous pour ajouter HyperaiXingXing sur WeChat (ID WeChat : Hyperai01) et notez « SD Tutorial Exchange Group » pour rejoindre le chat de groupe.










