Command Palette
Search for a command to run...
LeCun a Transmis, UC Berkeley Et al. a Proposé Une Méthode De Génération De Protéines Multimodale PLAID, Qui Génère Des Séquences Et Des Structures Protéiques Entièrement Atomiques En Même Temps

Au cours des dernières années, les scientifiques ont continué à explorer la structure et la composition des protéines afin de mieux démêler le « code de la vie ».La fonction des protéines est déterminée par leur structure, y compris l’identité et la position des atomes de la chaîne latérale et de la chaîne principale et leurs propriétés biophysiques, qui sont collectivement appelées la structure entièrement atomique.Cependant, pour déterminer où placer les atomes de la chaîne latérale, il faut d’abord connaître la séquence. Par conséquent, la génération de structures entièrement atomiques peut être considérée comme un problème multimodal qui nécessite la génération simultanée de séquence et de structure.
Cependant, les méthodes existantes de génération de structure et de séquence de protéines traitent généralement la séquence et la structure comme des modes indépendants. Les méthodes de génération de structure génèrent généralement uniquement des atomes de la chaîne principale. Les méthodes ciblant la conception de tous les atomes nécessitent généralement l'utilisation de modèles externes pour alterner entre la prédiction de structure et les étapes d'anti-pliage, etc.
Pour relever ces défis, une équipe de recherche de l'Université de Californie à Berkeley (UC Berkeley), de Microsoft Research et de Genentech a proposé une méthode de génération de protéines multimodales appelée PLAID (Protein Latent Induced Diffusion), qui peut réaliser une génération multimodale en cartographiant des modalités de données plus riches (telles que des séquences) vers des modalités plus rares (telles que des structures cristallines).Pour valider l’approche, les chercheurs ont mené des expériences sur 2 219 fonctions de l’ontologie génétique et 3 617 organismes à travers l’arbre de la vie.Même si aucune entrée structurelle n'est utilisée pendant la formation, les échantillons générés présentent une forte qualité structurelle et une forte cohérence.
La recherche connexe s'intitule « Génération de structures protéiques tout-atome à partir de données d'entraînement de séquence uniquement » et a été soumise à la conférence de haut niveau ICLR 2025. Yang Likun, le « Parrain de l'IA », a également republié cette réalisation sur la plateforme sociale.
Adresse open source du projet PLAID :
http://github.com/amyxlu/plaid
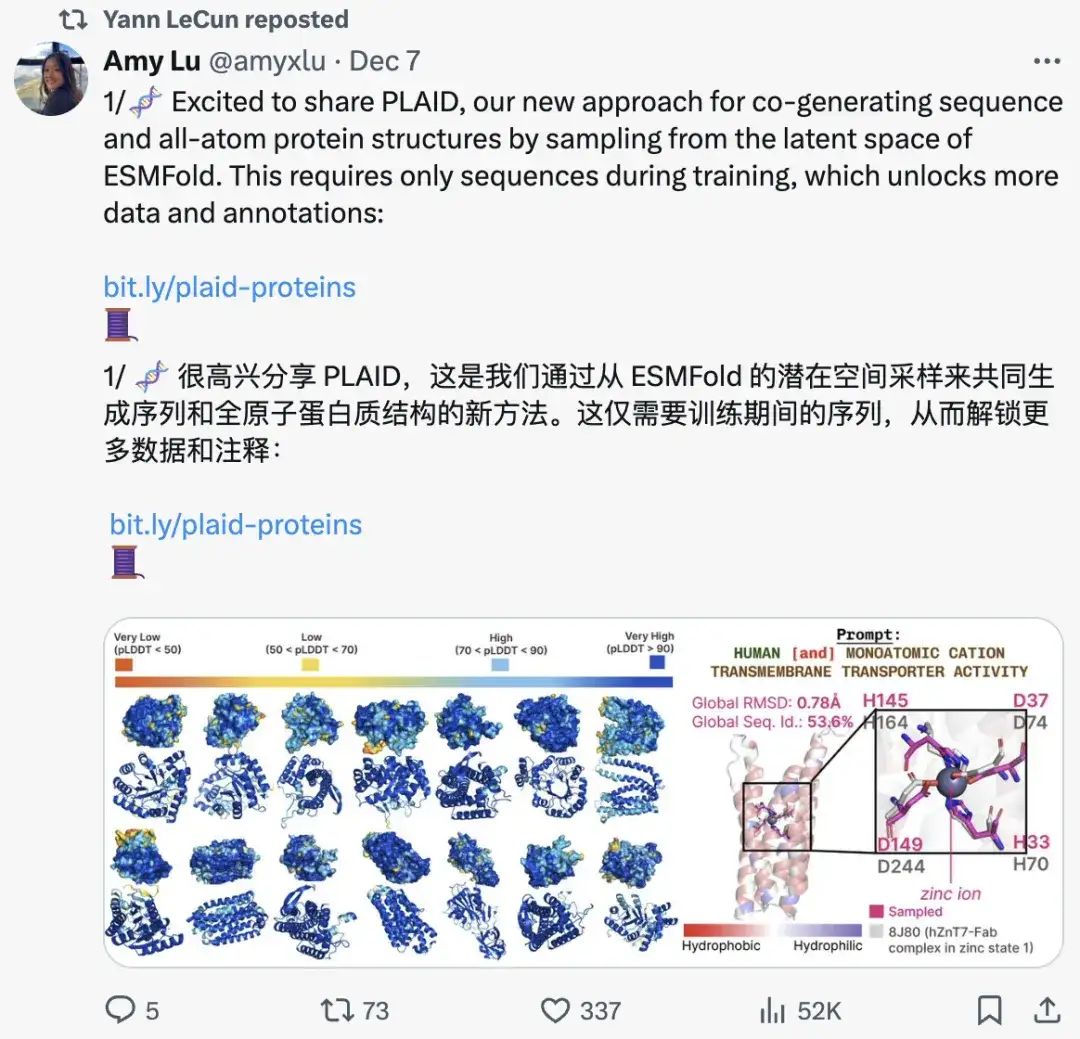
Points saillants de la recherche :
* En se concentrant sur le grand modèle de langage protéique ESMFold et la génération de structures entièrement atomiques, les chercheurs ont proposé un modèle de diffusion contrôlable qui peut générer simultanément des séquences et des structures protéiques entièrement atomiques, ne nécessitant qu'une entrée de séquence pendant la formation.
* L’approche exploite les informations structurelles codées dans les poids pré-entraînés plutôt que dans les données d’entraînement et augmente la disponibilité des annotations de séquence pour une génération contrôlable.
* Bien que le modèle ESMFold soit utilisé dans l’article, la méthode peut être appliquée à n’importe quel modèle de prévision.
Adresse du document :
https://www.biorxiv.org/content/10.1101/2024.12.02.626353v1
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Un aperçu rapide des points saillants de la recherche
Ensemble de données
Les chercheurs ont utilisé la version de septembre 2023 de la base de données Pfam, qui contient 57 595 205 séquences et 20 795 familles. PLAID est entièrement compatible avec des bases de données de séquences plus grandes telles que UniRef ou BFD (environ 2 milliards de séquences), cependant cette étude a choisi d'utiliser Pfam car son domaine de séquence contient plus de balises structurelles et fonctionnelles, ce qui rend l'évaluation par simulation informatique des échantillons générés plus pratique. De plus, les chercheurs ont conservé environ 15% de données à des fins de vérification.
Les codes UniRef des organismes à partir desquels les domaines Pfam ont été dérivés sont disponibles à partir du fichier Pfam-A.fasta fourni sur le serveur FTP Pfam. Les chercheurs ont analysé tous les organismes uniques de l’ensemble de données, trouvant un total de 3 617 organismes différents, puis ont mené des expériences sur ces organismes pour vérifier l’efficacité de la méthode PLAID.
Architecture du modèle
PLAID est un nouveau paradigme pour la génération multimodale et contrôlable de protéines par diffusion dans l'espace latent des modèles prédictifs.L'aperçu de la méthode est présenté dans la figure ci-dessous. En bref, il se divise en 4 étapes :
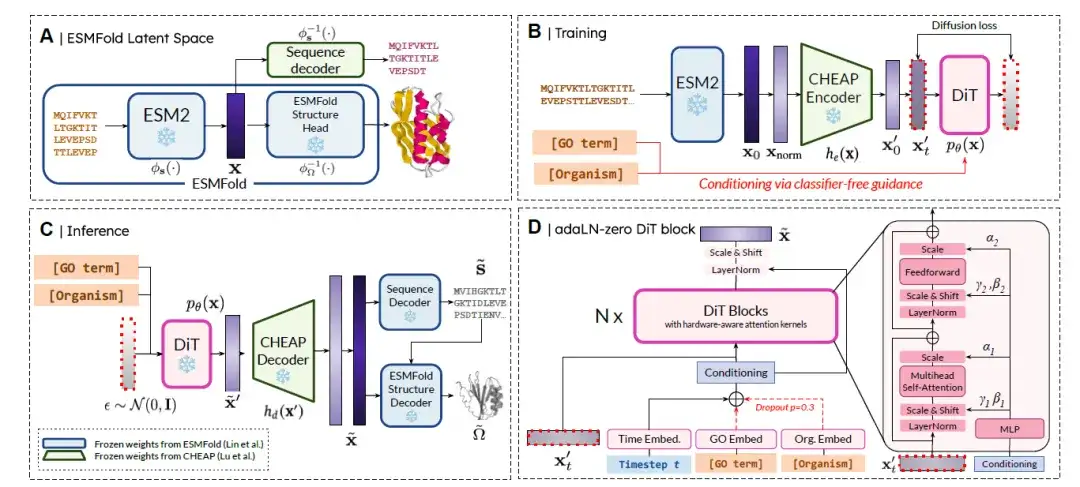
(A) Espace latent ESMFold :L'espace latent p(x) représente l'intégration conjointe de la séquence et de la structure.
(B) Formation à la diffusion potentielle :L'objectif est d'apprendre et d'échantillonner à partir de pθ(x), en suivant la formule de diffusion. Pour améliorer l'efficacité de l'apprentissage, les chercheurs utilisent l'encodeur CHEAP he(·) pour obtenir l'intégration compressée x′ = he(x), de sorte que l'objectif de diffusion devient l'échantillonnage à partir de pθ(he(x)).
(C) Inférence :Pour capturer à la fois la séquence et la structure au moment de l'inférence, nous utilisons le modèle formé pour échantillonner ˜x′ ∼ pθ(x′), puis le décompressons à l'aide du décodeur CHEAP pour obtenir ˜x = hd(˜x′). L'intégration est décodée dans la séquence d'acides aminés correspondante par un décodeur de séquence congelée formé dans CHEAP. La séquence d'identité des résidus et ˜x sont utilisés comme entrée dans un décodeur de structure gelée formé dans ESMFold pour obtenir la structure de tous les atomes.
(D) Architecture du bloc DiT :Les chercheurs ont utilisé l'architecture Diffused Transformer (DiT) combinée au bloc DiT adaLN-zero pour fusionner les informations conditionnelles. Les étiquettes fonctionnelles (c'est-à-dire les termes GO) et les étiquettes de classe d'organisme ont été intégrées sans aucune orientation de classificateur.
Résultats de l'étude
Les chercheurs ont effectué une analyse de la qualité structurelle et de la diversité de différentes longueurs de protéines, et les résultats sont présentés dans la figure ci-dessous.Les échantillons de protéines natives et générés par PLAID ont des métriques cohérentes à différentes longueurs.ProteinGenerator et Protpardelle ont montré un effondrement du mode à certaines longueurs, tandis que Multiflow a montré une diversité diminuée sur des séquences plus longues.
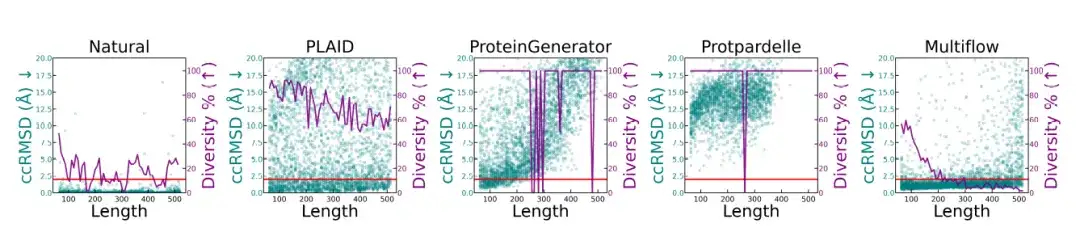
* Cette figure compare les protéines naturelles et différentes méthodes de génération, montrant la qualité structurelle (ccRMSD, points cyan) et la diversité (ligne violette, mesurée comme la proportion de groupes structurels uniques dans l'échantillon total) des protéines à différentes longueurs (64-512 résidus). La ligne rouge est à 2 Å, indiquant le seuil de conception)
De plus, par rapport à la méthode de référence,La diversité de la structure secondaire générée par PLAID ressemble davantage à la distribution des protéines natives.Comme le montre la figure ci-dessous : ProteinGenerator, Protpardelle et Multiflow présentent des écarts dans leurs distributions de structures secondaires, et les modèles de génération de structures protéiques existants ont généralement du mal à générer des échantillons avec une teneur élevée en feuillets β.
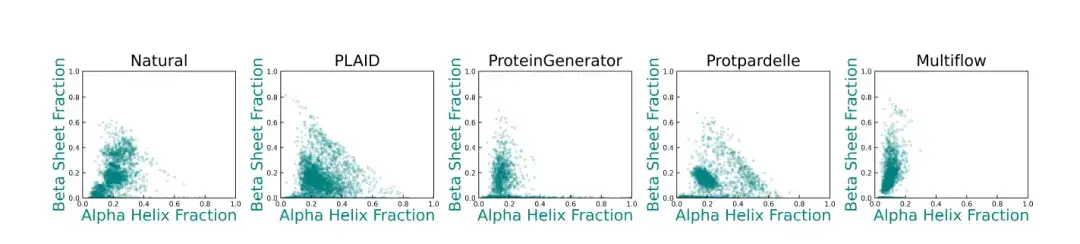
* La figure montre la distribution de la teneur en hélices α et en feuillets β des protéines naturelles et des structures protéiques générées par différentes méthodes. Chaque point représente une structure et ses coordonnées représentent la proportion de résidus hélicoïdaux α (axe des x) et la proportion de résidus de feuillets β (axe des y).
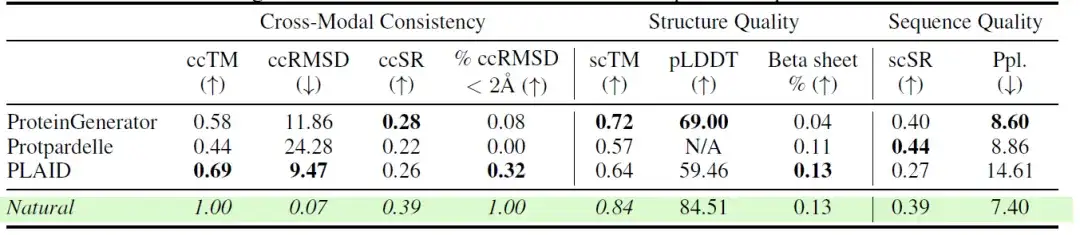
Les chercheurs ont également comparé les performances de différents modèles sur plusieurs mesures de cohérence et de qualité dans la tâche de génération de protéines entièrement atomiques. Les résultats sont présentés dans le tableau suivant :Les échantillons générés par PLAID montrent une cohérence intermodale élevée entre la séquence et la structure.
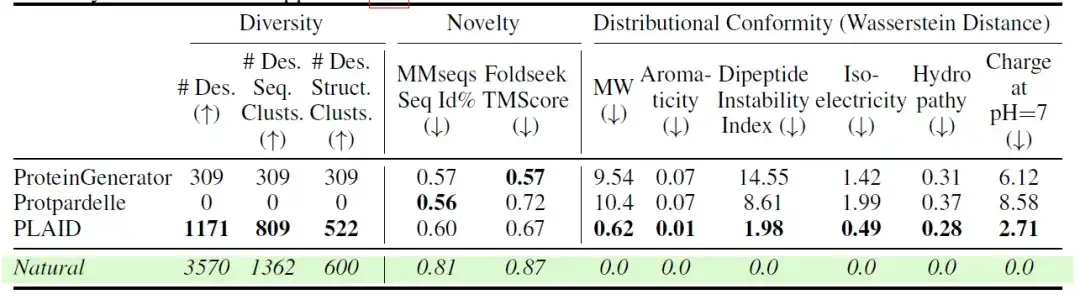
Les chercheurs ont également évalué la diversité, la nouveauté et le caractère naturel des différents modèles. Les résultats sont présentés dans le tableau suivant :Parmi les modèles entièrement atomiques, PLAID a généré les échantillons les plus uniques et les plus conçus, à la fois dans l'espace séquentiel et structurel.
Il convient de souligner que PLAID peut être facilement étendu à de nombreuses fonctions en aval et ne se limite pas à ESMFold, mais peut être appliqué à n'importe quel modèle de prédiction.
L'IA ouvre une nouvelle voie pour la recherche sur les protéines
Le transformateur de diffusion est de plus en plus utilisé dans le domaine biologique
Cet article mentionne que pendant le processus de construction du modèle, les chercheurs ont utilisé un transformateur de diffusion (DiT) pour effectuer des tâches de débruitage.
Le principe de base de DiT est d'appliquer l'architecture Transformer au modèle de diffusion. Les modèles de diffusion corrompent généralement les données d'origine en ajoutant progressivement du bruit, puis récupèrent ces données grâce à l'apprentissage du modèle. DiT améliore la capacité générative du modèle en introduisant des blocs Transformer (tels que la normalisation de couche adaptative, l'attention croisée, etc.) dans le modèle de diffusion.
Ces dernières années, DiT a réalisé des progrès significatifs dans le domaine de la génération d’images et de vidéos. L'architecture principale des modèles de génération de pointe tels que Sora est DiT.Dans le domaine de la biomédecine, l’application du transformateur de diffusion devient de plus en plus étendue. Il peut aider les chercheurs à sélectionner rapidement des molécules médicamenteuses potentielles et à prédire leur activité biologique. Il peut également aider à des tâches complexes telles que l’analyse de séquences génétiques et la prédiction de la structure des protéines, fournissant ainsi un outil puissant pour la recherche en sciences de la vie.Prenons l’exemple de la réduction du bruit des protéines, DiT peut capturer des relations séquence-structure complexes. Autrement dit, grâce au mécanisme d'auto-attention globale de Transformer, il peut modéliser efficacement la relation interactive complexe entre la séquence et la structure des protéines, puis utiliser le processus inverse du modèle de diffusion pour prédire le vecteur latent débruité à chaque pas de temps, restaurant progressivement la structure et la séquence de la protéine à partir du bruit.
Spécifiquement pour cet article, DiT fournit des options plus flexibles pour un réglage précis afin de gérer des modalités d'entrée mixtes, en particulier lorsque les modèles de prédiction de la structure des protéines commencent à intégrer les acides nucléiques et les complexes de ligands de petites molécules. De plus, cette approche permet une meilleure utilisation de l’infrastructure de formation Transformer.
Lors des premières expériences, les chercheurs ont également découvert que l’allocation de la mémoire disponible à des modèles DiT plus grands était plus efficace que l’utilisation de l’auto-attention triangulaire. Il utilise le modèle de formation d'algorithme d'optimisation implémenté par xFormers et a obtenu une amélioration de la vitesse de 55,8% et une réduction de 15,6% de l'utilisation de la mémoire GPU dans le test de référence de la phase d'inférence.
L'apprentissage automatique fait des protéines personnalisées un « rêve devenu réalité »
La recherche mentionnée ci-dessus de l’UC Berkeley peut être considérée comme une autre étape importante dans la personnalisation des protéines. Nous savons que les protéines sont généralement constituées de 20 acides aminés différents, qui peuvent être considérés comme les éléments constitutifs de la vie.En raison de sa structure extrêmement complexe, il y a quelques décennies, prédire la structure tridimensionnelle des protéines et concevoir de nouvelles protéines à usage humain était encore un « rêve irréaliste » pour les scientifiques. Cependant, les progrès rapides de l’apprentissage automatique ces dernières années ont rendu progressivement possible le rêve de concevoir des protéines personnalisées.
Outre le célèbre AlphaFold, certaines avancées de la recherche méritent également l’attention :
En novembre 2024, une équipe du laboratoire national d'Argonne du département américain de l'Énergie a développé avec succès un cadre informatique innovant appelé MProt-DPO.Le cadre combine la technologie de l’intelligence artificielle avec les meilleurs superordinateurs du monde, marquant une nouvelle ère dans la conception des protéines. En prenant MProt-DPO comme exemple, les scientifiques ont conçu un nouveau type d’enzyme capable de catalyser efficacement des réactions chimiques dans des conditions spécifiques. Par rapport aux méthodes de conception précédentes, l'efficacité de la nouvelle réaction enzymatique est améliorée de près de 30%, ce qui non seulement accélère les progrès expérimentaux, mais offre également davantage de possibilités pour les applications industrielles. De plus, l’application réussie du MProt-DPO ouvre également de nouvelles idées pour la conception de protéines antivirales. Les résultats de recherche pertinents ont été publiés dans l'IEEE Computer Society sous le titre « MProt-DPO : Breaking the ExaFLOPS Barrier for Multimodal Protein Design Workflows with Direct Preference Optimization ».
Adresse du document :
https://www.computer.org/csdl/proceedings-article/sc/2024/529100a074/21HUV88n1F6
Les poches protéiques sont des sites sur les protéines qui sont adaptés à la liaison à des molécules spécifiques. La conception de poches protéiques est l’une des méthodes importantes dans le processus de personnalisation des protéines. En décembre 2024, l'Université des sciences et technologies de Chine et ses collaborateurs ont conçu l'algorithme de génération profonde PocketGen.Des séquences et des structures de poches protéiques peuvent être générées sur la base de la structure protéique et des petites molécules liées. Les expériences montrent que les indicateurs tels que l'affinité du modèle PocketGen et la rationalité structurelle dépassent les méthodes traditionnelles, et l'efficacité de calcul est également grandement améliorée. Les résultats de recherche pertinents ont été publiés dans Nature Machine Intelligence sous le titre « Génération efficace de poches de protéines avec PocketGen ».
Adresse du document :
https://www.nature.com/articles/s42256-024-00920-9

À l’avenir, avec l’application plus poussée de l’intelligence artificielle dans le domaine des protéines, je pense que les gens auront une compréhension plus approfondie des secrets de la structure spatiale des protéines.
Références :
1.https://www.biorxiv.org/content/10.1101/2024.12.02.626353v1
2.https://mp.weixin.qq.com/s/_5_L7bvl-vHtls8gBbfSmQ
3.https://mp.weixin.qq.com/s/sfrm2rj_8kH0JA2vu4NmTw
4.http://www.news.cn/globe/20241014/f7137840e56340f081f9eb819d87ba40/c.html
5.http://www.bfse.cas.cn/yjjz/202412/t20241212_5042432.html
6.https://www.sohu.com/a/826241274_12








