Command Palette
Search for a command to run...
Publié Dans La Revue Nature ! L'Université De Californie Utilise l'IA Pour Innover Dans La Reconstruction 3D Par Cryomicroscopie Électronique Et Réalise Une Avancée Majeure En Biologie Structurale

Dans le domaine de la recherche scientifique, certaines technologies deviennent souvent le centre de l’attention en raison de leurs progrès révolutionnaires. La microscopie cryoélectronique (Cryo-EM), qui a remporté le prix Nobel de chimie 2017, est l’une de ces technologies. Par exemple, en s'appuyant sur la technologie de la cryomicroscopie électronique, l'équipe de Shi Yigong a capturé la structure haute résolution du spliceosome pour la première fois en 2015. Cela a été salué comme la plus grande contribution de la Chine à la science mondiale dans le domaine des sciences de la vie fondamentales au cours des 30 dernières années, et cela a également suscité une large attention pour la cryomicroscopie électronique.
En tant qu'outil important dans le domaine de la biologie structurale, la microscopie cryoélectronique peut refroidir rapidement les échantillons à basse température, empêchant la cristallisation des molécules d'eau dans les échantillons, préservant ainsi l'état quasi physiologique des échantillons.Une fois les échantillons congelés, les chercheurs peuvent utiliser une gamme de techniques cryo-EM pour visualiser les échantillons en 3D à différentes résolutions, y compris une résolution quasi atomique, afin d'obtenir une compréhension plus approfondie et plus complète des échantillons.
Cependant, bien que la technologie de la microscopie cryoélectronique soit devenue de plus en plus mature, la question de l’avantage d’orientation lors de la préparation des échantillons a toujours été un problème difficile. En général, le processus de reconstruction 3D nécessite des projections de protéines dans toutes les directions pour couvrir tout l’espace. Cependant, les protéines adsorbées à l'interface air-liquide (AWI) présentent souvent des avantages d'orientation, ce qui entraîne des ensembles de données de projection incomplets, qui à leur tour provoquent divers degrés de distorsion de la densité protéique, conduisant à des distorsions de reconstruction.
récemment,L'équipe de recherche de l'UCLA a proposé une méthode d'apprentissage profond auto-supervisée appelée IsoNet à particule unique (spIsoNet).Cette méthode fournit une nouvelle façon de restaurer l’isotropie des échantillons. Lorsque spIsoNet est appliqué à la cryo-EM à particules uniques, il peut améliorer considérablement la qualité de la reconstruction des macromolécules biologiques, améliorer la précision de l'alignement et l'isotropie angulaire et apporter de nouvelles avancées dans le domaine de la biologie structurale.
La recherche, intitulée « Surmonter le problème de l'orientation préférée dans la cryo-EM avec l'apprentissage profond auto-supervisé », a été publiée dans la revue universitaire internationale Nature Methods.
Points saillants de la recherche :
* Cette étude a développé une méthode de bout en bout basée sur l'apprentissage profond auto-supervisé, spIsoNet, qui peut être utilisée pour améliorer la qualité d'image de la cryo-EM
* spIsoNet peut résoudre le problème de reconstruction 3D causé par le problème d'orientation des préférences
* spIsoNet améliore l'isotropie angulaire et la précision de l'alignement des particules lors de la reconstruction 3D

Adresse du document :
https://doi.org/10.1038/s41592-024-02505-1
Adresse du jeu de données spIsoNet :
https://go.hyper.ai/P7XQu
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : sélectionnez plusieurs ensembles de données, chacun avec des caractéristiques et des scénarios d’application différents
Dans cette étude, les chercheurs ont utilisé plusieurs ensembles de données pour tester les performances de spIsoNet, chacun avec ses propres caractéristiques et scénarios d'application uniques :
* Ensemble de données sur la β-galactosidase :Il contient deux sous-ensembles avec des orientations spécifiques, à savoir 1 513 particules en vue latérale et 950 particules en vue de dessus, qui sont utilisées pour vérifier si spIsoNet peut améliorer la qualité de l'image (qualité de la carte) affectée par l'orientation préférée.
* Ensemble de données inclinées du trimère HA (EMPIAR-10097) :Elle est obtenue grâce à une stratégie d'inclinaison de grille, qui fournit une direction de visualisation inclinée et peut être utilisée pour évaluer la capacité de spIsoNet à gérer des échantillons inclinés.
* Ensemble de données de trimère HA non asymétrique (EMPIAR-10096) :L'image a été collectée dans des conditions d'inclinaison sans grille et, en important 130 000 particules et en effectuant une correction de désalignement, l'image résultante avait une résolution de 3,45 Å, ce qui peut être utilisé pour comparer les différences d'effets de traitement entre les échantillons inclinés et non inclinés.
* Ensemble de données sur les ribosomes asymétriques (EMPIAR-10406) :Il contient le ribosome 70S du pathogène A. baumannii complexé avec l'amikacine et a été utilisé pour évaluer les performances de spIsoNet dans le traitement de structures biomoléculaires complexes.
* Ensemble de données de tomographie VLP du VIH (EMPIAR-10164) :Il contient des particules de type virus VIH-1 dMACANC immatures (VLP) à une résolution de 3,6 Å. Dans cette étude, nous avons fourni un aperçu approfondi de la structure de la particule virale.
spIsoNet : Basé sur le modèle d'apprentissage profond U-net, il se compose de deux modules principaux
Le réseau neuronal utilisé par spIsoNet est basé sur l'architecture réseau U-net.Il s’agit d’un modèle d’apprentissage profond qui a acquis une large reconnaissance dans la restauration et la segmentation d’images biomédicales. Comme le montre la figure b ci-dessous, U-net est construit sur la base de l'architecture encodeur-décodeur en empilant des blocs convolutifs.
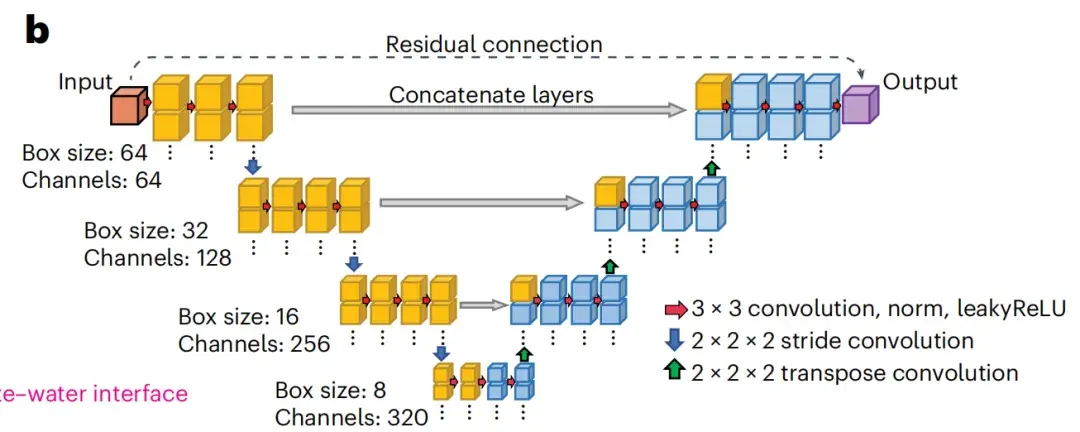
Basé sur le modèle U-net, spIsoNet se compose principalement de deux modules :
Le module de correction d'anisotropie
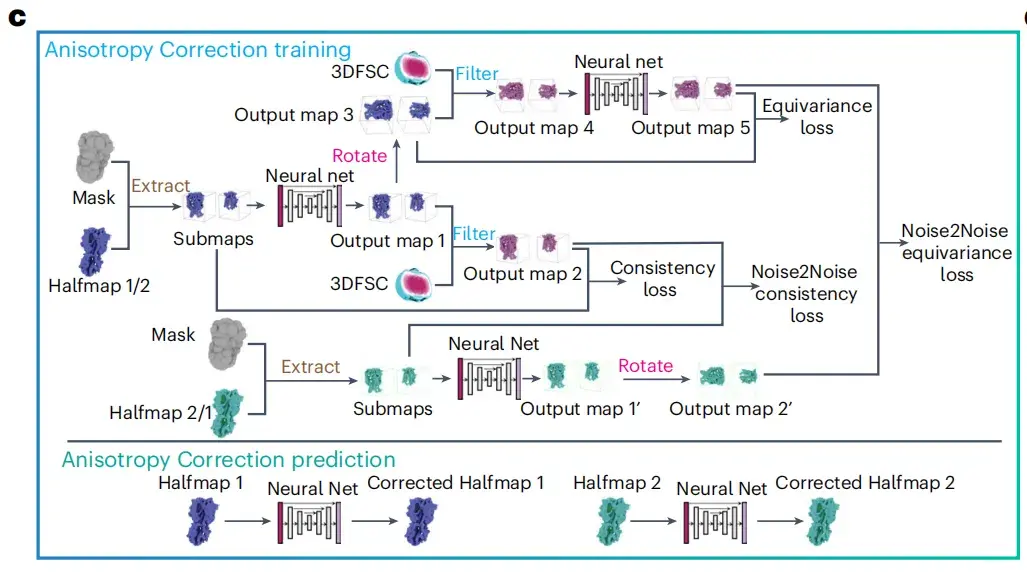
Les chercheurs ont conçu un module de correction d’anisotropie pour améliorer la clarté des images de microscopie cryoélectronique.Comme le montre la figure c ci-dessous, le fonctionnement de ce module prend deux demi-cartes, un volume de corrélation de coque de Fourier tridimensionnelle (3DFSC) et un masque de solvant comme données d'entrée, et intègre l'algorithme 3DFSC pour minimiser la somme pondérée de quatre types différents de fonctions de perte, y compris la perte de cohérence, la perte d'équivariance, la perte de cohérence bruit à bruit et la perte d'équivariance bruit à bruit, améliorant ainsi la qualité des images cryo-EM.
Le module de correction du désalignement alimenté par la correction d'anisotropie
Comme le montre la figure e ci-dessous, ce module intègre un flux de travail comprenant trois étapes principales : filtrage de carte, correction d'anisotropie et affinage automatique RELION. La correction de l’anisotropie est au cœur de l’ensemble du processus, qui vise à améliorer la qualité des images cryo-EM grâce à la correction de l’anisotropie.
* La correction d'anisotropie fait référence à la correction des différences dans les propriétés physiques et chimiques d'un objet dans différentes directions grâce à des algorithmes spécifiques pour obtenir un effet isotrope.
* La technologie de correction du désalignement est principalement utilisée pour corriger les problèmes de désalignement de l'image causés par la distorsion géométrique pendant le processus d'imagerie.
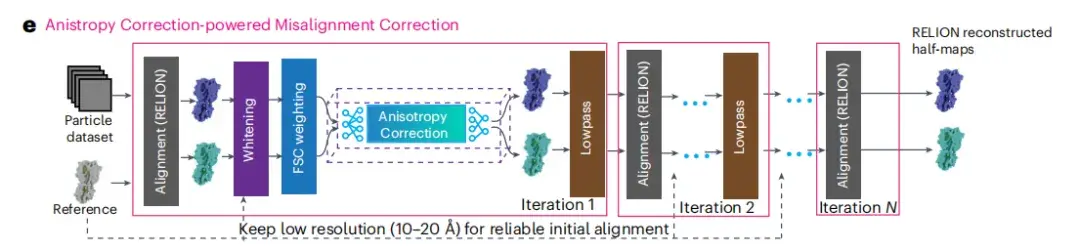
Après avoir terminé la correction d'anisotropie, les chercheurs ont obtenu des paramètres d'orientation des particules plus précis et deux demi-images reconstruites par RELION. Après chaque itération de raffinement 3D, ces demi-images sont traitées via des filtres de post-traitement incluant le blanchiment et la pondération FSC pour améliorer encore la qualité de l'image. Le module de correction anisotrope spIsoNet traite ensuite ces demi-images filtrées, et les demi-images corrigées traitées sont filtrées passe-bas pour atteindre une norme correspondant à leur résolution. Ces deux demi-images filtrées et corrigées serviront de références pour l'estimation ultérieure de l'orientation.
Résultats de recherche : spIsoNet améliore considérablement la qualité des images cryo-EM
La correction de l'anisotropie est efficace
Les chercheurs ont découvert que le module de correction d’anisotropie de spIsoNet peut récupérer efficacement les informations manquantes dans les données simulées. donc,L’étude a d’abord testé spIsoNet sur l’ensemble de données du didacticiel RELION contenant la β-galactosidase.
Comme le montre la figure jm ci-dessous, en sélectionnant des particules vues latérales et des particules vues de dessus à partir des moyennes de classe 2D, les chercheurs ont compilé deux sous-ensembles de particules avec des orientations préférées et ont effectué une reconstruction 3D RELION standard. Les résultats des tests montrent que le module de correction d'anisotropie seul peut réduire efficacement les artefacts de reconstruction 3D causés par les orientations dominantes en vue de dessus ou en vue latérale.
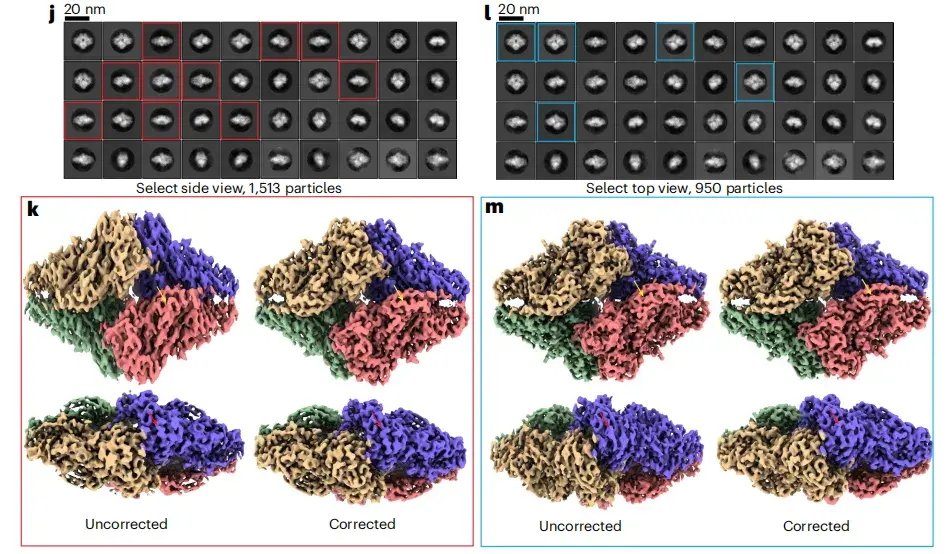
* Où jl fait référence à la carte de classification 2D reconstruite sous différents angles, et km fait référence à la carte de classification 3D reconstruite sous différents angles.
Les techniques de correction de l'anisotropie et de correction du désalignement améliorent considérablement la qualité de l'image cryo-EM
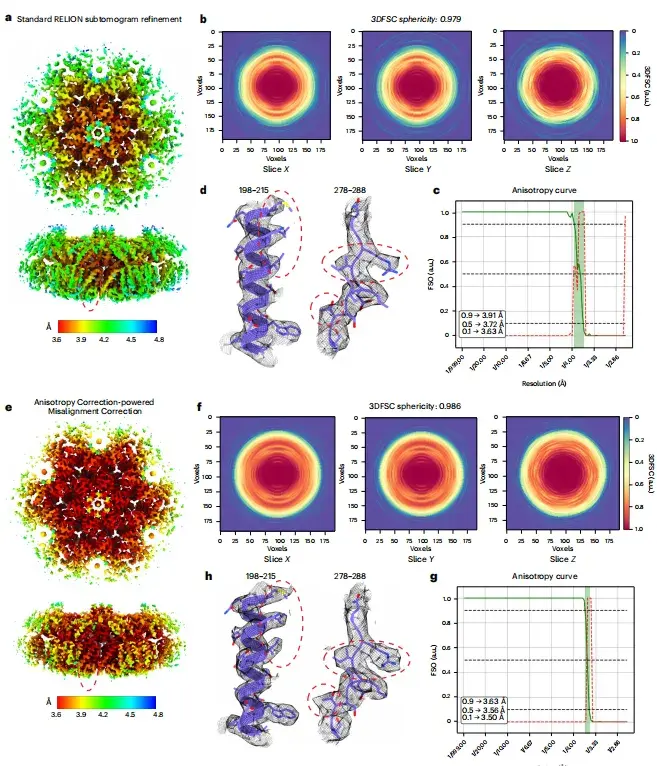
Des études antérieures ont montré que la qualité de l’image cryo-EM des ensembles de données inclinés du trimère HA n’est pas idéale. Afin de tester l'effet de spIsoNet,Cette étude a d’abord effectué une correction anisotrope sur la demi-carte.Les résultats montrent que la qualité de l’image corrigée est considérablement améliorée, la résolution locale est augmentée et le bruit est réduit. Comme le montrent les figures ab ci-dessous, dans l’image corrigée, la densité de la chaîne latérale qui était auparavant difficile à discerner dans l’image d’origine devient clairement visible.
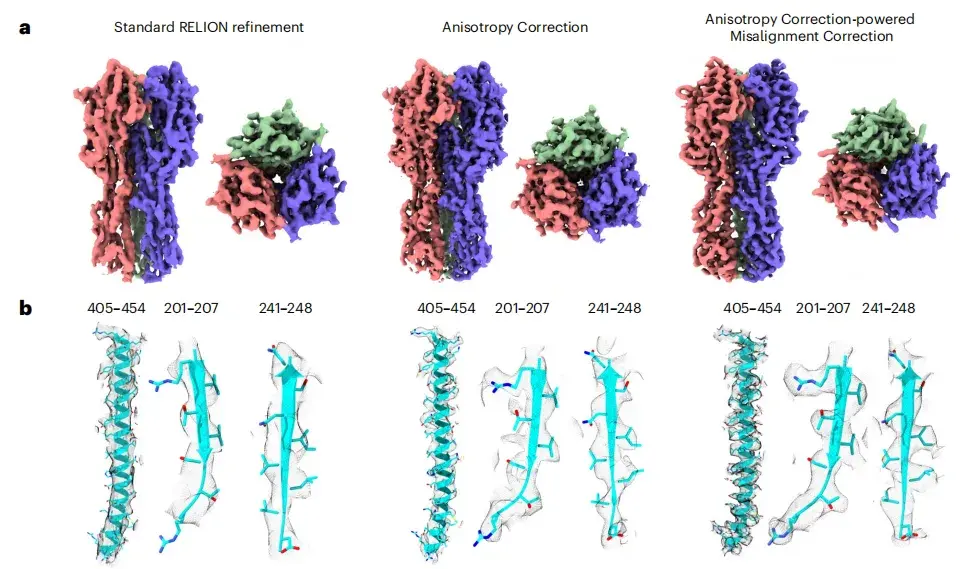
* De gauche à droite : raffinement RELION standard, correction d'anisotropie et correction de désalignement pilotée par la correction d'anisotropie.
De plus, comme le montre la figure cf ci-dessous, l'image après correction de déflexion améliore la corrélation de la coque de Fourier de l'image au modèle, et la corrélation de la coque de Fourier tridimensionnelle (3DFSC) est proche de la sphère (0,991). Par rapport à l'image originale, l'image après correction montre également une zone occupée par la couche de Fourier isotrope (FSO) plus grande.
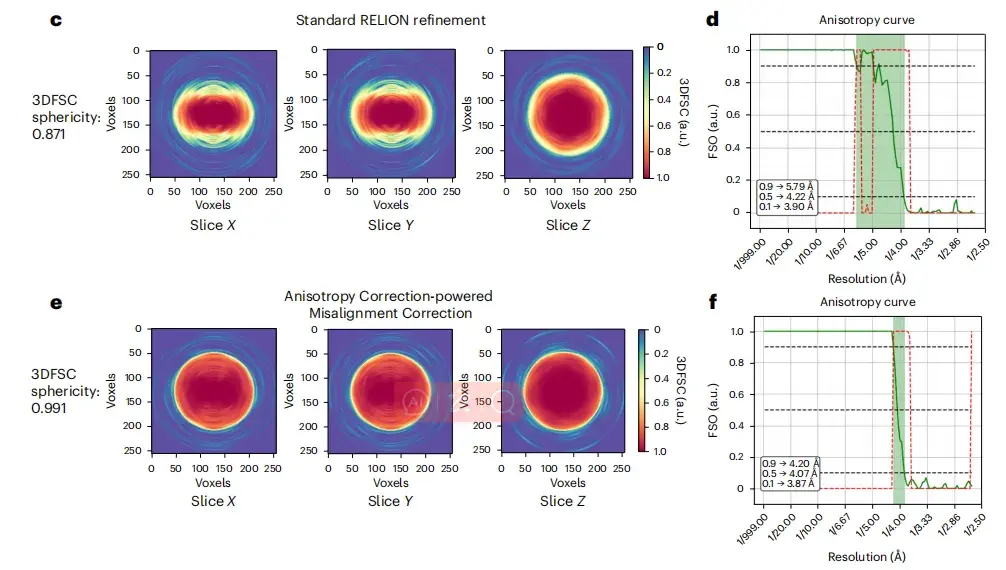
* Où, ce sont les tranches 3DFSC utilisées pour le raffinement RELION et la correction du désalignement spIsoNet, respectivement, et df sont les valeurs FSO et P du test de Bingham calculées à partir des résultats du raffinement RELION et de la correction du désalignement spIsoNet, respectivement.
La correction du désalignement a permis d'identifier et de corriger avec succès de nombreuses directions mal attribuées
Pour un ensemble de données protéiques présentant de sérieux problèmes d'orientation de biais, l'ensemble de données de trimère HA non incliné (EMPIAR-10096)Cette étude a utilisé le module de correction de désalignement piloté par correction d'anisotropie de spIsoNet pour traiter les ensembles de données de particules.L'image du trimère HA reconstruite dans l'ensemble de données incliné a été utilisée comme modèle de référence.
Après correction du désalignement, comme le montrent les figures b à f ci-dessous, les chercheurs ont obtenu une image avec la forme correcte et ont obtenu une amélioration significative de l'isotropie. Comme le montre la figure h ci-dessous, la résolution d'image déterminée par le FSC demi-carte (3,5 Å) et le FSC modèle-carte (3,6 Å) est cohérente.
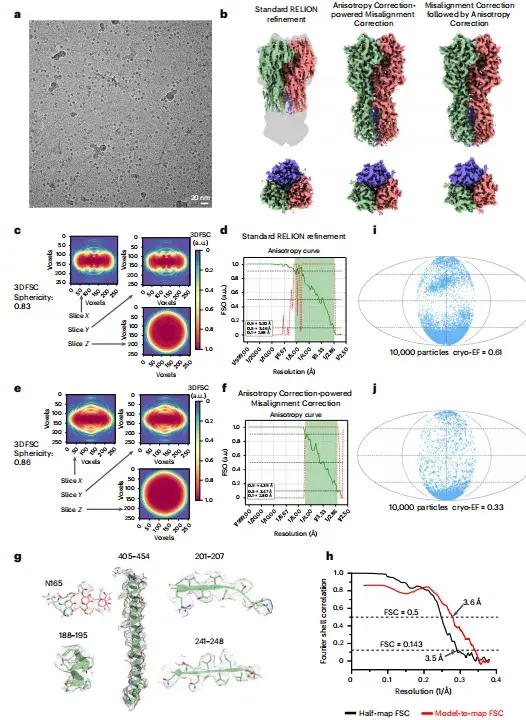
*a-micrographies cryo-EM représentatives, b-images cryo-EM du trimère HA reconstruites par différentes méthodes, c-tranches de 3DFSC utilisées pour le raffinement standard RELION, d-FSO et valeurs P du test de Bingham calculées sur la base des résultats du raffinement standard RELION, e-tranches de 3DFSC corrigées par le désalignement spIsoNet, f-FSO et valeurs P du test de Bingham calculées sur la base des résultats de correction du désalignement spIsoNet, g-densité représentative des résidus d'acides aminés et des glycanes sélectionnés à partir des images cryo-EM, h-courbe FSC corrigée du trimère HA, i,j-résultats de distribution dans différentes directions et scores cryoEF correspondants
spIsoNet excelle dans l'amélioration de l'alignement des particules asymétriques et des particules contenant des molécules d'acide nucléique
Comme le montrent les figures ci-dessous, après correction de l'anisotropie, la qualité de l'image est considérablement améliorée, montrant une distribution de densité plus continue, une résolution locale plus élevée et moins d'interférences de bruit.L'étude a révélé queLorsque la moyenne du sous-tomogramme local des ribosomes 70S ou 80S est utilisée comme référence et que la résolution initiale de 15 Å est maintenue pour l'alignement, des images de haute qualité sans biais de modèle peuvent être obtenues de manière cohérente et les effets de l'anisotropie peuvent être efficacement atténués.
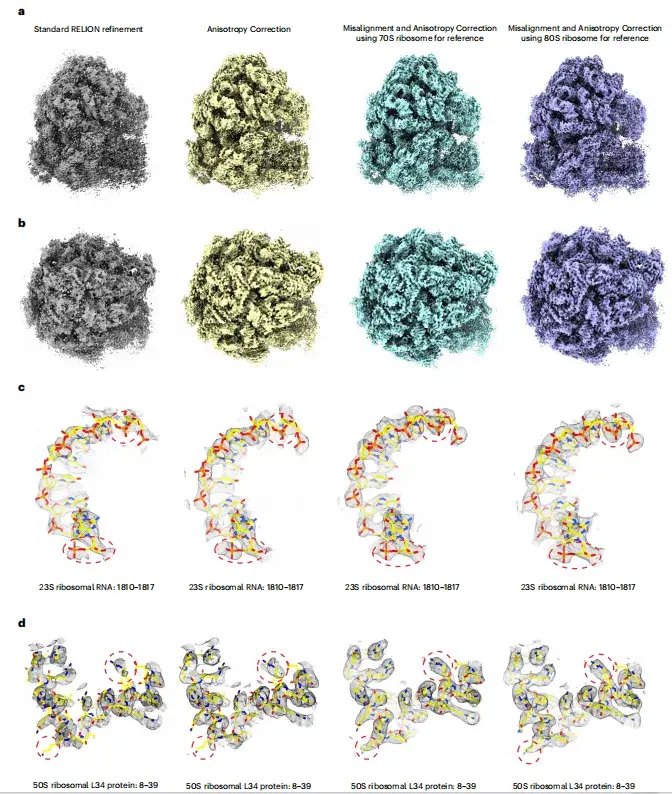
* a, b- images de ribosomes reconstruites par différentes méthodes de reconstruction, c, d- régions de densité représentatives avec des modèles atomiques ajustés (jaune)
spIsoNet a un potentiel d'application en biologie structurale in situ
Pour évaluer l’application de spIsoNet dans la moyenne des sous-tomogrammes, cette étude a utilisé l’ensemble de données de tomographie VLP du VIH-1 (EMPIAR-10164) comme exemple.
Comme le montre la figure a ci-dessous, dans le processus standard de RELION4, cette étude a utilisé un sous-ensemble de 5 groupes d'angles d'inclinaison différents pour obtenir une structure avec une résolution de 3,7 Å. Ensuite, en appliquant une correction de désalignement, les chercheurs ont obtenu une structure isotrope de résolution 3,6 Å comme le montre la figure e ci-dessous.
Comme le montrent les figures bh ci-dessous, l'analyse structurelle révèle en outre une densité de chaîne latérale plus claire et présente une sphéricité 3DFSC plus élevée dans la courbe FSO, ce qui contribue à améliorer la précision de l'estimation de l'orientation des particules.
* a- Carte de résolution locale du VIH-1 reconstruite selon la norme RELION, b- tranche de 3DFSC utilisée pour le raffinement standard RELION, c- FSO et valeur P du test de Bingham calculés selon les résultats du raffinement standard RELION, d- densité représentative des résidus d'acides aminés et des glycanes sélectionnés à partir d'images cryo-EM, e- Carte de résolution locale du VIH-1 reconstruite selon la technologie de correction d'anisotropie spIsoNet, f- tranche de 3DFSC corrigée par l'anisotropie spIsoNet, g- FSO et valeur P du test de Bingham calculés selon les résultats de la correction d'anisotropie spIsoNet, h- densité représentative des résidus d'acides aminés et des glycanes sélectionnés à partir d'images cryo-EM
IA + cryomicroscopie électronique, un paradigme technologique de « combinaison forte »
Au cours des deux dernières années, un sujet controversé a fait surface dans la communauté scientifique : « AlphaFold met-il fin à la biologie structurale ? » La réponse est bien sûr non.
d'une part,Les données de formation pour les modèles de prédiction de structure tels qu'AlphaFold proviennent de méthodes d'analyse de structure traditionnelles telles que les rayons X et la microscopie cryoélectronique.d'autre part,La microscopie cryoélectronique excelle dans l’analyse de la dynamique des protéines, ce qui n’est actuellement pas possible avec AlphaFold. Alors, la technologie d’IA représentée par AlphaFold peut-elle aider les méthodes traditionnelles représentées par la cryomicroscopie électronique ? On peut dire que c’est une nécessité.
Par exemple, dès 2022,L'équipe du professeur Mao Youdong de l'Université de Pékin a utilisé l'IA + la cryomicroscopie électronique pourLa conversion transitoire du protéasome induit d'un état intermédiaire de dégradation du substrat à un état intermédiaire d'inhibition du substrat a été capturée avec succès. C'est la première fois au monde qu'une technologie de reconstruction quadridimensionnelle par intelligence artificielle est appliquée pour améliorer la précision analytique de la microscopie cryoélectronique à résolution temporelle. L'équipe a réalisé une observation de la dynamique fonctionnelle au niveau atomique de complexes protéiques cibles liés à des maladies majeures. Les résultats associés ont été publiés dans Nature sous le titre « Allostérie régulée par USP14 du protéasome humain par cryo-EM résolu dans le temps ».
Lien vers l'article :
https://doi.org/10.1038/s41586-022-04671-8
Il n'y a pas longtemps,Les chercheurs de l’équipe de recherche ByteDance ont proposé une nouvelle méthode appelée CryoSTAR.Il s’agit de la première méthode permettant d’appliquer les priors modaux de structure atomique des protéines aux données expérimentales de cryomicroscopie électronique. Il utilise les informations du modèle atomique comme régularisation structurelle pour élucider l'hétérogénéité conformationnelle des macromolécules biologiques. Il peut produire des modèles à gros grains et des cartes de densité pour montrer les changements conformationnels des molécules à différents niveaux, améliorant considérablement le potentiel d'application de la microscopie cryoélectronique dans l'analyse conformationnelle dynamique. Les résultats associés ont été publiés dans Nature Methods sous le titre « CryoSTAR : Leveraging Structural Prior and Constraints for Cryo-EM Heterogeneous Reconstruction ».
Lien vers l'article :
https://www.nature.com/articles/s41592-024-02486-1
Il ne fait aucun doute que la combinaison de l’IA et de la cryomicroscopie électronique ouvre un nouveau chapitre de la biologie structurale et démontre également le grand potentiel de la technologie de l’IA pour aider les méthodes traditionnelles de biologie structurale.








