Command Palette
Search for a command to run...
Publié Dans La Revue Science ! L'Université Jiao Tong De Shanghai Et Le Shanghai AI Lab Ont Publié Conjointement Un Modèle De Conception De Mutant Protéique Qui Surpasse Les Méthodes Les Plus Avancées

Les protéines ne sont pas seulement l'exécuteur des activités de la vie humaine, mais jouent également un rôle important dans de nombreux domaines tels que la biomédecine, la transformation des aliments, l'industrie brassicole, l'industrie chimique, etc. Par conséquent, les gens n'ont jamais cessé de mener des recherches sur la structure des protéines, la fonction, etc., pour sélectionner des protéines qui répondent aux besoins et sont très stables pour les scénarios d'application industrielle.
Cependant, les conditions physiques et chimiques (telles que la température et le pH) nécessaires pour que les protéines « de type sauvage » extraites d’organismes fonctionnent dans des environnements industriels sont pour la plupart éloignées de leur environnement biologique natif. En d’autres termes, la stabilité de ce type de protéine rend difficile son adaptation aux environnements industriels difficiles. Par conséquent, afin de répondre aux besoins de différents scénarios d’application,Des mutations sont souvent nécessaires pour améliorer les propriétés physicochimiques des protéines, augmentant ainsi leur stabilité dans des conditions de température/pH extrêmes ou augmentant l'activité et la spécificité des enzymes.
Il convient de noter que la modification de l’activité biologique d’une protéine nécessite des années de recherche expérimentale sur son mécanisme de fonctionnement, ce qui est non seulement long et laborieux, mais aussi de plus en plus difficile à répondre aux besoins de modification en constante évolution. Ces dernières années, l’émergence de modèles de langage protéique a considérablement amélioré la précision de la prédiction de la forme physique des protéines, mais elle manque encore de précision dans la prédiction de la stabilité.
Les mutations protéiques véritablement significatives devraient améliorer la stabilité tout en maintenant leur activité biologique, et vice versa. En réponse à cela, le groupe de recherche du professeur Hong Liang à l'École des sciences naturelles/École de physique et d'astronomie de l'Université Jiao Tong de Shanghai, en collaboration avec Tan Pan, un jeune chercheur au Laboratoire d'intelligence artificielle de Shanghai, et des collaborateurs de l'Université ShanghaiTech et de l'Académie chinoise des sciences du Collège médical de Hangzhou,Ils ont développé conjointement une nouvelle méthode de pré-formation de modèle de langage à grande échelle de séquences protéiques PRIME,Dans le même temps, les meilleurs résultats de prédiction ont été obtenus dans la prédiction de l’activité de mutation des protéines et de la stabilité des mutations, ainsi que dans d’autres apprentissages de représentation liés à la température.
La recherche connexe, intitulée « Un modèle général de langage guidé par la température pour concevoir des protéines de stabilité et d'activité améliorées », a été publiée dans Science Advances, une revue bien connue de Science.
Points saillants de la recherche :
* PRIME peut prédire l'amélioration des performances de mutants protéiques spécifiques sans s'appuyer sur des données expérimentales antérieures
* PRIME peut prédire efficacement plusieurs propriétés d'une protéine, permettant aux chercheurs de concevoir avec succès des protéines dans des domaines inconnus
* PRIME est formé sur la base d'un modèle de langage « sensible à la température », qui peut mieux capturer les caractéristiques de température des séquences de protéines

Adresse du document :
https://www.science.org/doi/10.1126/sciadv.adr2641
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : 96 millions d'enregistrements, explorant la relation entre la séquence protéique et la température
En intégrant les données publiques d'Uniprot (Universal Protein Resource) et les séquences protéiques obtenues à partir d'échantillons environnementaux grâce à des études métagénomiques,Des chercheurs ont compilé une grande base de données, ProteomeAtlas, contenant 4,7 milliards de séquences de protéines naturelles.
* UniProt est une grande base de données fournissant des séquences de protéines et des annotations détaillées associées.
Au cours du processus de sélection des séquences, les chercheurs ont conservé uniquement les séquences complètes et ont traité ces séquences à l'aide de l'outil d'alignement de séquences biologiques MMseqs2, en définissant le seuil d'identité de séquence à 50% pour réduire la redondance, puis en identifiant et en annotant les séquences liées aux températures de croissance optimales (OGT) des souches bactériennes.
final,Les chercheurs ont ainsi annoté 96 millions de séquences protéiques.Il fournit une ressource riche pour explorer la relation entre la séquence protéique et la température.
De plus, dans l'analyse de la capacité prédictive à zéro tir de la stabilité thermique du modèle, les ensembles de données utilisés pour étudier le changement de température de fusion (ΔTm) ont été dérivés de MPTherm, FireProtDB et ProThermDB, et toutes les expériences ont été réalisées dans les mêmes conditions de pH.
Parmi eux, MPTherm contient des données expérimentales liées à la stabilité thermique des protéines ; FireProtDB est spécifiquement utilisé pour stocker des données expérimentales de mutation liées à la stabilité thermique et à la fonction des protéines ; ProThermDB collecte spécifiquement des données liées aux propriétés thermodynamiques des protéines. Dans le même temps, les chercheurs ont également combiné des données d’analyse des mutations profondes (DMS), provenant principalement de la base de données d’analyse des mutations protéiques ProteinGym.
* Ensemble de données sur les mutations protéiques ProteinGym
https://go.hyper.ai/YlMT5
Architecture du modèle : Modèle d'apprentissage profond basé sur la « perception de la température »
Le nouveau modèle d'apprentissage profond PRIME (Protein language model for Intelligent Masked pretraining and Environment prediction) proposé par l'institut,Capacité à prédire les améliorations de performances de mutants protéiques spécifiques sans s'appuyer sur des données expérimentales antérieures.
Le modèle est formé sur la base d'un modèle de langage « sensible à la température », s'appuyant sur un ensemble de données de 96 millions de séquences protéiques, combinant la tâche de modélisation du langage masqué (MLM) au niveau du jeton et la cible de prédiction de la température de croissance optimale (OGT) au niveau de la séquence, et introduisant le terme de perte de corrélation via l'apprentissage multitâche. Il peut filtrer les séquences protéiques présentant une tolérance à haute température pour optimiser leur stabilité et leur activité biologique.
Spécifiquement,PRIME se compose de 3 parties principales :Comme le montre la figure ci-dessous. Le premier est le module Encoder, qui est un encodeur Transformer utilisé pour extraire les caractéristiques latentes de la séquence. Le deuxième est le module MLM, conçu pour aider l'encodeur à apprendre la représentation contextuelle des acides aminés. Dans le même temps, le module MLM peut également être utilisé pour la notation des mutants. Le troisième composant est le module de prédiction OGT, qui peut prédire l'OGT de l'organisme dans lequel la protéine est située en fonction de la représentation potentielle.
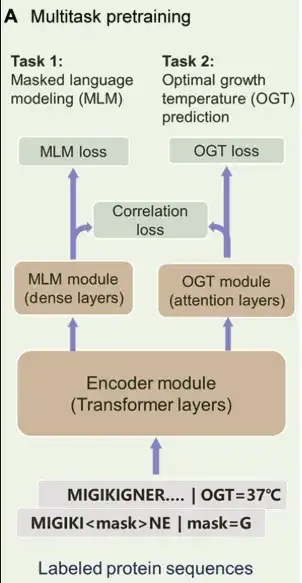
L'apprentissage multitâche de PRIME dans la phase de pré-formation comprend le MLM, la prédiction OGT et la perte de corrélation.
dans,Le MLM est souvent utilisé comme méthode de pré-formation pour la représentation des données de séquence.Dans cette étude, des séquences protéiques bruyantes ont été utilisées comme entrée, certaines étiquettes ont été masquées ou représentées par des étiquettes alternatives, et l'objectif de la formation était de reconstruire ces étiquettes bruyantes. Cette approche aide le modèle à capturer les dépendances entre les acides aminés et les informations contextuelles de la séquence, tout en utilisant ce processus de reconstruction pour noter les mutations.
La deuxième tâche d’entraînement a été optimisée dans des conditions supervisées et les chercheurs ont utilisé un ensemble de données de 96 millions de séquences protéiques annotées avec OGT pour entraîner le modèle PRIME. L'entrée de cette tâche est une séquence protéique et les valeurs de température générées par le module OGT vont de 0° à 100°C. Il convient de noter que le module OGT et le module MLM fonctionnent à l'aide d'un encodeur partagé.Cette structure permet au modèle de capturer simultanément les informations contextuelles des acides aminés ainsi que les caractéristiques de séquence dépendantes de la température.
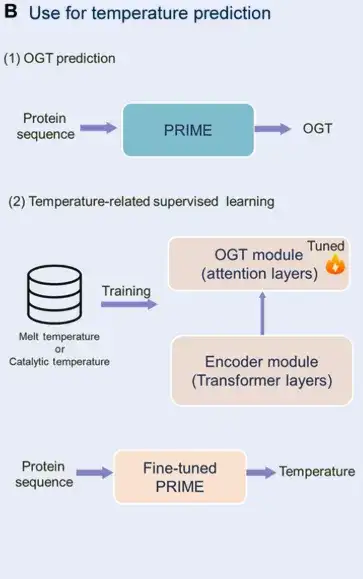
Enfin, les chercheurs ont introduit la perte de corrélation pour faciliter la rétroaction de la classification OGT prédite à la classification MLM, en alignant les informations sur les tâches aux niveaux du jeton et de la séquence.Cela permet au grand modèle de mieux capturer les caractéristiques de température des séquences de protéines.
Conclusion expérimentale : surpasse les méthodes les plus avancées pour prédire l'adaptabilité des séquences de protéines mutantes
Les chercheurs ont comparé expérimentalement les capacités de prédiction à zéro tir de PRIME avec celles des modèles les plus avancés de stabilité thermique, notamment les modèles d'apprentissage profond ESM-1v, ESM-2, MSA-transformer, Tranception-EVE, CARP, MIF-ST, SaProt, Stability Oracle et les méthodes de calcul traditionnelles GEMME et Rosetta.
Les chercheurs ont utilisé des ensembles de données de MPTherm, FireProtDB et ProThermDB, qui contiennent des changements de température de fusion (ΔTm) collectés dans le même environnement de pH et ont assuré qu'il y avait au moins 10 points de données pour chaque protéine, pour un total de 66 détections. L’étude a également intégré un test de balayage de mutation profonde (DMS), en utilisant ProteinGym comme banc d’essai.
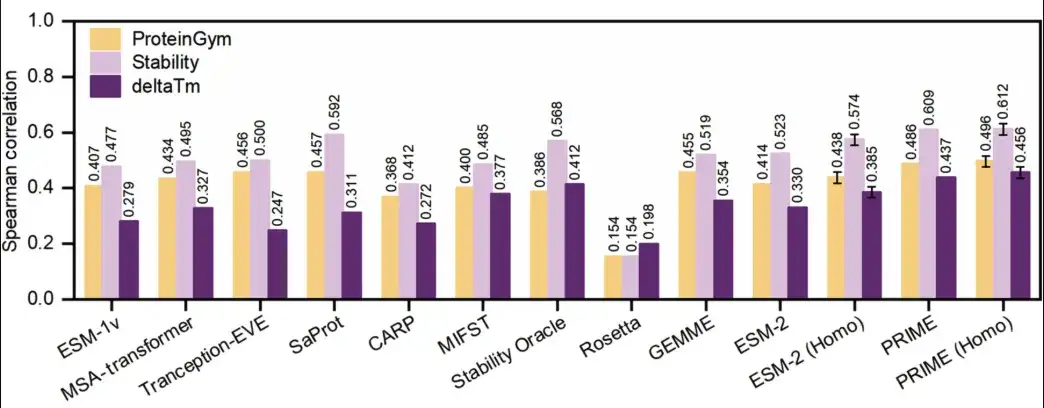
Les résultats sont présentés dans la figure ci-dessous.PRIME surpasse toutes les autres méthodes pour prédire à la fois la disponibilité et la stabilité des protéines.
Dans le benchmark ProteinGym (jaune dans la figure ci-dessous), PRIME a obtenu un score de 0,486 et le deuxième SaProt a obtenu un score de 0,457. Dans l'ensemble de données ΔTm (violet foncé dans la figure ci-dessous), PRIME occupe toujours la première place avec un score de 0,437, et la deuxième place a un score de 0,412. De plus, les chercheurs ont également comparé PRIME avec d’autres méthodes du sous-ensemble de données de stabilité ProteinGym (violet clair dans la figure ci-dessous), et PRIME a toujours surpassé toutes les autres méthodes.
Il convient de noter que pour tester l’efficacité et l’effet de PRIME dans l’application pratique de l’ingénierie des protéines,Les chercheurs ont également mené une expérience humide et sélectionné cinq protéines à des fins de vérification.Comprend LbCas12a, l'ARN polymérase T7, la créatinase, l'acide nucléique polymérase artificielle et la région variable de la chaîne lourde d'un nanoanticorps spécifique.
Dans les tests expérimentaux des 30 à 45 principales mutations ponctuelles, plus de 30% des mutants ponctuels recommandés par l'IA étaient significativement supérieurs aux protéines de type sauvage dans des propriétés clés telles que la stabilité thermique, l'activité enzymatique, l'affinité de liaison antigène-anticorps, la capacité de polymérisation des acides nucléiques non naturels ou la tolérance dans des conditions alcalines extrêmes, et le taux positif des protéines individuelles dépassait 50%.
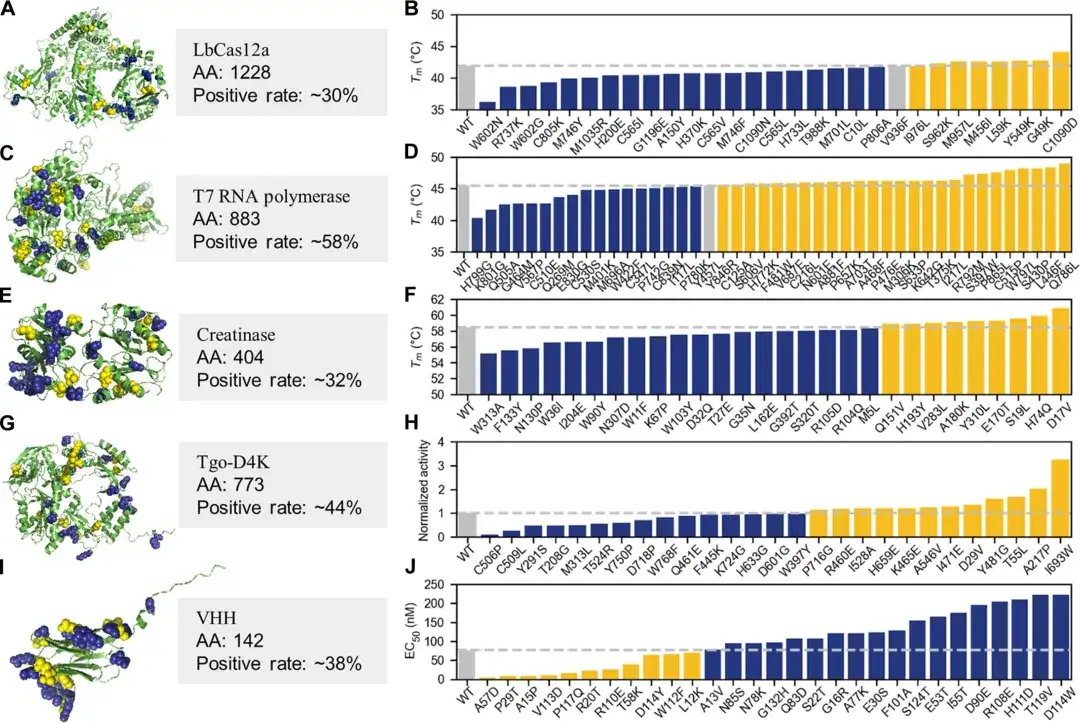
Il convient de mentionner que l’équipe a également démontré une méthode efficace basée sur PRIME.Des mutants multisites avec une activité et une stabilité améliorées peuvent être rapidement obtenus.Grâce à cette méthode de réglage fin sur de petits échantillons, d'excellents mutants protéiques peuvent être produits en 2 à 4 cycles d'évolution avec moins de 100 échantillons expérimentaux humides.
Par exemple, après quatre séries d’itérations sèches-humides, l’ARN polymérase T7 a réussi à obtenir des mutants multipoints avec une activité élevée et une stabilité élevée. Le mutant multipoint le plus élevé avait une Tm de 12,8 °C supérieure à celle du type sauvage et son activité était près de quatre fois supérieure à celle du type sauvage. Les performances de certains produits ont dépassé celles de produits similaires qui ont dominé le marché pendant 10 ans par une société internationale de biotechnologie de premier plan (New England Biolabs). De plus, dans les expériences de LbCas12a et de l'ARN polymérase T7, Pro-PRIME peut superposer des mutations ponctuelles négatives pour obtenir des mutations ponctuelles positives.
Cela montre que PRIME peut apprendre les effets épistatiques des mutations protéiques à partir de données de séquence, ce qui est d'une grande importance pour l'ingénierie protéique traditionnelle.
Approfondir l'ingénierie des protéines pour surmonter le problème des petits échantillons
Dans le domaine de l’ingénierie des protéines, l’expression, la purification et les tests fonctionnels des protéines nécessitent généralement des réactifs et des instruments coûteux, et les expériences prennent du temps, ce qui limite considérablement le nombre d’échantillons pouvant être générés. Dans la recherche sur la fonction des protéines, tester les effets des mutations protéiques sur les fonctions (telles que l'activité catalytique, la stabilité thermique, l'affinité de liaison, etc.) nécessite des expériences plus précises et plus complexes, et il est difficile de mesurer les performances de toutes les mutations possibles de manière unique et à haut débit.
Cela rend difficile pour les modèles d’apprentissage automatique d’obtenir une formation suffisante sur des échantillons limités, ce qui entraîne de mauvaises performances du modèle dans la prédiction de nouvelles mutations. De plus, les erreurs expérimentales ou le bruit dans les données de petits échantillons peuvent provoquer une plus grande interférence avec la formation du modèle. On peut dire queLe défi posé par les petits échantillons de données a, dans une certaine mesure, limité l’efficacité et la précision de la recherche dans le domaine de l’ingénierie des protéines.Cela a grandement motivé les chercheurs à explorer des technologies innovantes, combinant l’apprentissage automatique, les techniques expérimentales et l’analyse de données multimodales pour dépasser les limites des petits échantillons.
L’équipe de recherche décrite dans cet article a réalisé des résultats remarquables à cet égard. En plus de PRIME mentionné ci-dessus, l'équipe du professeur Hong Liang et le Dr Tan Pan ont également publié un certain nombre de résultats sur l'apprentissage sur de petits échantillons.
Auparavant, l’équipe utilisait une combinaison d’apprentissage par méta-transfert (MTL), d’apprentissage par classement (LTR) et de réglage fin efficace des paramètres (PEFT).Nous avons développé une stratégie de formation, FSFP, qui peut optimiser efficacement les modèles de langage protéique lorsque les données sont extrêmement rares.Il peut être utilisé pour l’apprentissage sur de petits échantillons de l’adaptabilité des protéines. Il améliore considérablement l'effet des grands modèles traditionnels de pré-entraînement des protéines dans la prédiction des propriétés de mutation lorsqu'on utilise très peu de données expérimentales humides, et montre également un grand potentiel dans les applications pratiques.
La recherche connexe a été intitulée « Améliorer l'efficacité des modèles de langage protéique avec un minimum de données de laboratoire humide grâce à un apprentissage en quelques coups » et publiée dans Nature Communications, une filiale de Nature.
En outre, le professeur Hong Liang a également partagé des points de vue pertinents. Il estime que « dans les trois prochaines années, dans les domaines de la conception des protéines, du développement de médicaments, du diagnostic des maladies, de la découverte de nouvelles cibles, de la conception de voies de synthèse chimique et de la conception de matériaux, l'intelligence artificielle générale dans les domaines professionnels entraînera un changement de paradigme clair, transformant le modèle de découverte scientifique qui reposait sur des essais et des erreurs sporadiques du cerveau humain dans le passé en un modèle de conception standard automatisé à grand modèle d'IA. »
Les changements spécifiques incluent la création de méthodes d’apprentissage à échantillon nul ou à petit échantillon et la création de modèles technologiques de pré-formation.En l'absence de données, une grande quantité de fausses données avec une précision légèrement inférieure est générée via un simulateur physique pour la pré-formation, puis affinée avec des données réelles et précieuses pour compléter l'apprentissage par renforcement.
Le professeur Hong a souligné que « les fausses données désignent des données qui ne proviennent pas du monde réel, mais qui présentent un certain degré de fiabilité. Elles peuvent être générées par l'IA ou obtenues par simulation physique pour améliorer les données. Enfin, les données expérimentales réelles en milieu humide sont les plus précieuses et servent à affiner le modèle. »
En effet, le défi de la rareté des données ne se limite pas au domaine de l’ingénierie des protéines. Les méthodes d’apprentissage à petits échantillons, voire à échantillons nuls, sont cruciales. Nous attendons avec impatience que l’équipe du professeur Hong Liang et le Dr Tan Pan apportent davantage de résultats de haute qualité autour de ce point sensible.