Command Palette
Search for a command to run...
Publié Dans La Revue Nature ! L'Université Normale De Chine Centrale a Proposé DigFrag, Qui Utilise l'IA Pour Segmenter Avec Précision Des Fragments Moléculaires Et Générer 44 Molécules De médicaments/pesticides

Au cours des dernières décennies, la découverte de médicaments basée sur des fragments (FBDD) a joué un rôle important dans la recherche et le développement de nouveaux médicaments en identifiant de petits fragments de molécules qui ont de faibles interactions avec les protéines cibles et en optimisant les informations structurelles de ces fragments pour développer des composés principaux plus actifs.
Bien que le FBDD joue un rôle clé dans la découverte et le développement de médicaments, la construction et le criblage de bibliothèques de fragments moléculaires efficaces ont toujours été un défi majeur dans ce domaine. Les méthodes FBDD traditionnelles s’appuient sur l’intuition empirique, ce qui limite leur capacité à développer des structures diverses. Heureusement, l’émergence de l’IA apporte une solution transformatrice à ce défi.
Récemment, l'équipe du professeur Yang Guangfu et du professeur associé Wang Fan de l'Université normale de Chine centrale a développé une méthode de segmentation numérique appelée DigFrag.La méthode se concentre localement sur le graphe moléculaire, met en évidence les sous-structures clés et divise ces sous-structures en fragments. Les résultats expérimentaux montrent que les fragments segmentés par DigFrag présentent une diversité structurelle plus élevée et que les composés générés à partir de ces fragments sont plus cohérents avec les propriétés chimiques attendues. Cela suggère que les données générées à l’aide de méthodes d’IA peuvent être plus adaptées à la formation et à l’application de modèles d’IA.
La recherche, intitulée « DigFrag comme méthode de fragmentation numérique utilisée pour la conception de médicaments basée sur l'intelligence artificielle », a été publiée dans la revue universitaire internationale Nature Communications Chemistry.
Points saillants de la recherche :
* L'étude a révélé que lorsque des fragments basés sur DigFrag sont combinés avec des modèles d'IA, ils peuvent générer efficacement des molécules avec les propriétés souhaitées
* Grâce à un criblage précis, l’étude a finalement identifié 24 molécules médicamenteuses et 20 molécules pesticides
* L'équipe a développé une plateforme conviviale, MolFrag, qui intègre plusieurs technologies de fragmentation pour prendre en charge une plus large gamme de travaux d'analyse et de conception moléculaires
Adresse du document :
https://doi.org/10.1038/s42004-024-01346-5
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : base de données PADFrag auto-construite, contenant près de 3 000 types de données sur les médicaments
L'ensemble de données de modélisation utilisé dans cette étude provient principalement de la base de données auto-construite PADFrag. Plus précisément, la base de données PADFrag comprend principalement le catalogue de médicaments approuvé par la FDA dans la base de données DrugBank, qui comprend 1 652 médicaments, et les pesticides commerciaux répertoriés par Alan Wood, pour un total de 1 259.
*PADFrag, une base de données conçue pour explorer l'espace des fragments bioactifs pour la découverte de médicaments
https://pubs.acs.org/doi/10.1021/acs.jcim.8b00285
Afin de garantir la cohérence et la fiabilité des données, l’équipe de recherche a exclu les composés ayant des structures non standard. Par la suite, l'ensemble de données a été divisé en un ensemble d'entraînement, un ensemble de validation et un ensemble de test dans un rapport de 8:1:1 pour faciliter l'entraînement, l'évaluation et les tests du modèle.
DigFrag : un flux de travail en 3 étapes pour obtenir des fragments avec une plus grande diversité structurelle
DigFrag est une méthode de segmentation numérique innovante qui utilise un mécanisme d'attention graphique pour identifier et segmenter les fragments de médicaments/pesticides. Son principal avantage est qu’il peut obtenir des fragments avec une plus grande diversité structurelle du point de vue de l’intelligence artificielle plutôt que de s’appuyer uniquement sur l’expertise humaine.
De plus, l'étude a intégré les fragments segmentés par quatre méthodes, BRICS, RECAP, MacFrag et DigFrag, et les a intégrés dans le cadre du modèle DeepFMPO pour générer des molécules médicamenteuses et évaluer leurs performances sur différents indicateurs.
Enfin, en s'appuyant sur de multiples technologies de fragmentation moléculaire, les chercheurs ont développé une plateforme conviviale MolFrag pour soutenir le travail de segmentation moléculaire.
Plus précisément, le déroulement de cette étude est divisé en trois parties :
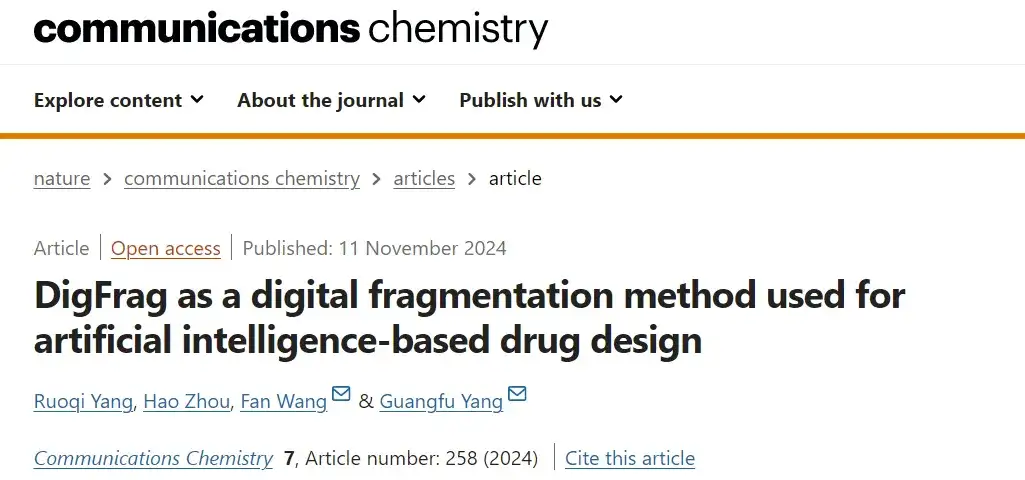
Tout d’abord, l’approche de fragmentation basée sur l’IA :Cette étude est basée sur l'architecture Graph Neural Network (GNN) et utilise la méthode DigFrag pour fragmenter les molécules.
Comme le montre la figure A ci-dessus, les chercheurs ont défini le graphique moléculaire comme G=(V, E), où V représente les nœuds, correspondant aux atomes de la molécule, et E représente les arêtes de connexion, correspondant aux liaisons chimiques entre les atomes. Dans ce processus, basé sur le réseau d'extraction de caractéristiques (matrice de caractéristiques) du mécanisme d'attention du graphe, le graphe moléculaire d'origine est d'abord introduit dans une série de couches d'attention pour obtenir une représentation d'intégration distincte pour chaque atome. Ces intégrations atomiques sont ensuite agrégées pour former un vecteur unifié, également appelé super nœud. Enfin, grâce à un traitement supplémentaire de la couche d'attention, la représentation intégrée du fragment entier est obtenue.
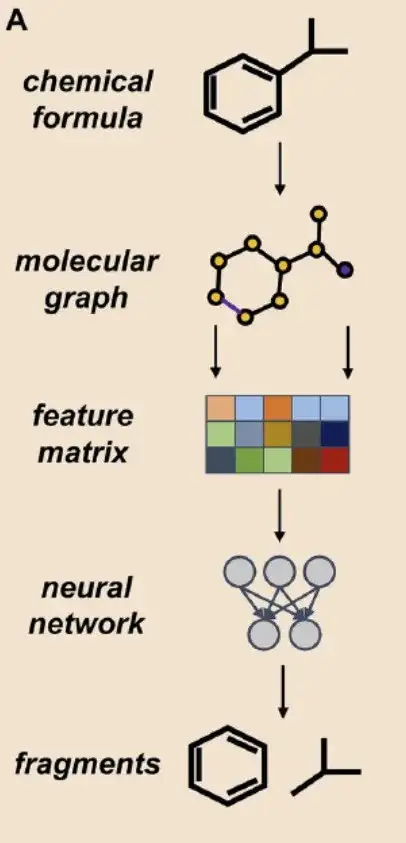
Deuxièmement, le cadre du modèle Acteur-CritiqueComme le montre la figure B ci-dessous, afin de clarifier davantage l'impact de la segmentation numérique sur les modèles génératifs profonds basés sur des fragments, les chercheurs ont intégré des fragments segmentés par quatre méthodes : BRICS, RECAP, MacFrag et DigFrag, et ont utilisé un outil open source de génération de molécules bidimensionnelles d'apprentissage par renforcement basé sur des fragments, l'architecture DeepFMPO, pour la recherche.
*DeepFMPO est un modèle d'apprentissage par renforcement acteur-critique qui obtient le composé souhaité en remplaçant des fragments dans le composé.
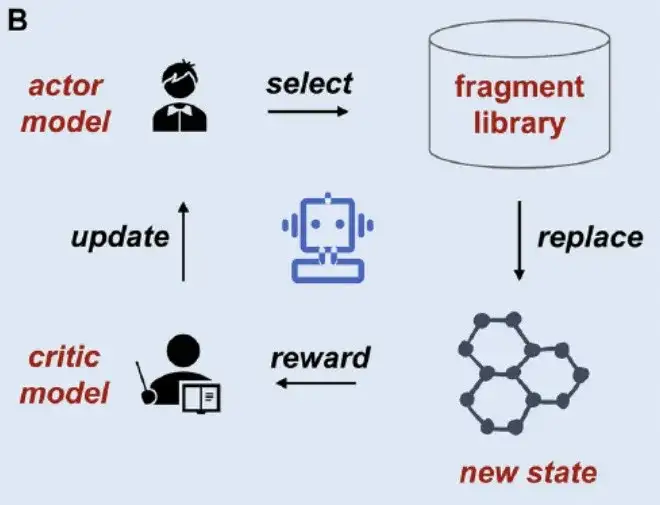
Troisièmement, établir une plateforme en ligne :Bien qu’une variété de méthodes de fragmentation moléculaire aient été développées, il existe un manque de serveurs en ligne faciles à utiliser. Par conséquent, comme le montre la figure C ci-dessus, cette étude a développé une plate-forme conviviale MolFrag basée sur diverses techniques de fragmentation. La plateforme combine de manière transparente quatre méthodes de fragmentation moléculaire : BRICS, RECAP, MacFrag et DigFrag, garantissant que les chercheurs de différents niveaux d'expertise peuvent l'utiliser.
Adresse de la plateforme MolFrag :
https://dpai.ccnu.edu.cn/MolFrag
Résultats de recherche : les fragments moléculaires segmentés DigFrag présentent une plus grande diversité
Les fragments DigFrag ont un grand nombre de liaisons rotatives
L’étude a d’abord formé le modèle à segmenter avec précision les fragments de médicaments et de pesticides. Ensuite, les chercheurs ont effectué une comparaison approfondie des trois indicateurs de performance clés de DigFrag, la précision du modèle, l'aire sous la courbe (AUC) et le coefficient de corrélation de Matthews (MCC) des fragments obtenus avec les méthodes traditionnelles (RECAP, BRICS) et les plus récentes (MacFrag) grâce à une validation croisée quintuple. Comme le montre le tableau ci-dessous, la distribution des propriétés des fragments de médicaments est plus similaire entre les fragments segmentés par DigFrag et ceux segmentés par BRICS.
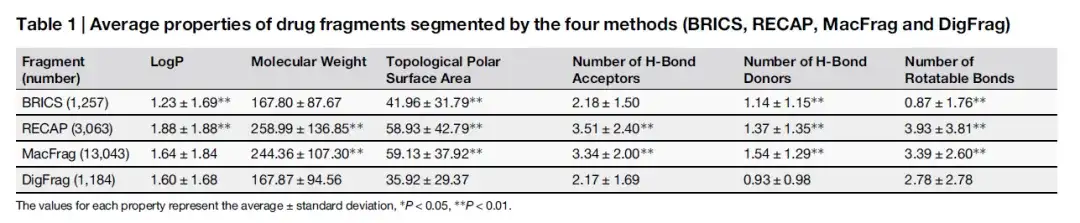
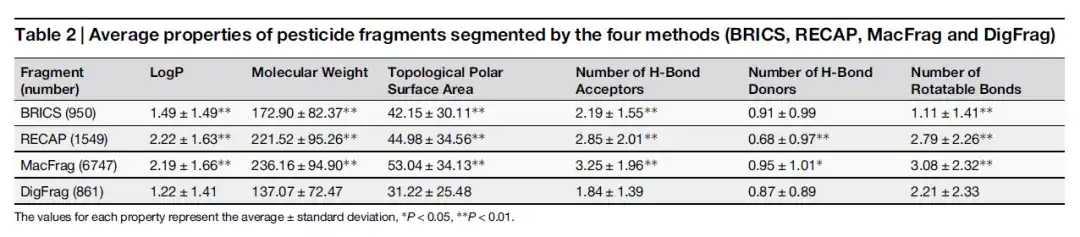
Comme le montre le tableau ci-dessous, bien que le poids moléculaire et le nombre d'accepteurs de liaisons H des fragments de médicament segmentés par DigFrag soient similaires à ceux segmentés par BRICS, le nombre de liaisons rotatives est plus grand, ce qui peut être lié à son mode de rupture de structure cyclique unique. En termes de fragments de pesticides, le poids moléculaire moyen des fragments segmentés par DigFrag est inférieur.
Les fragments segmentés DigFrag présentent une diversité structurelle plus élevée
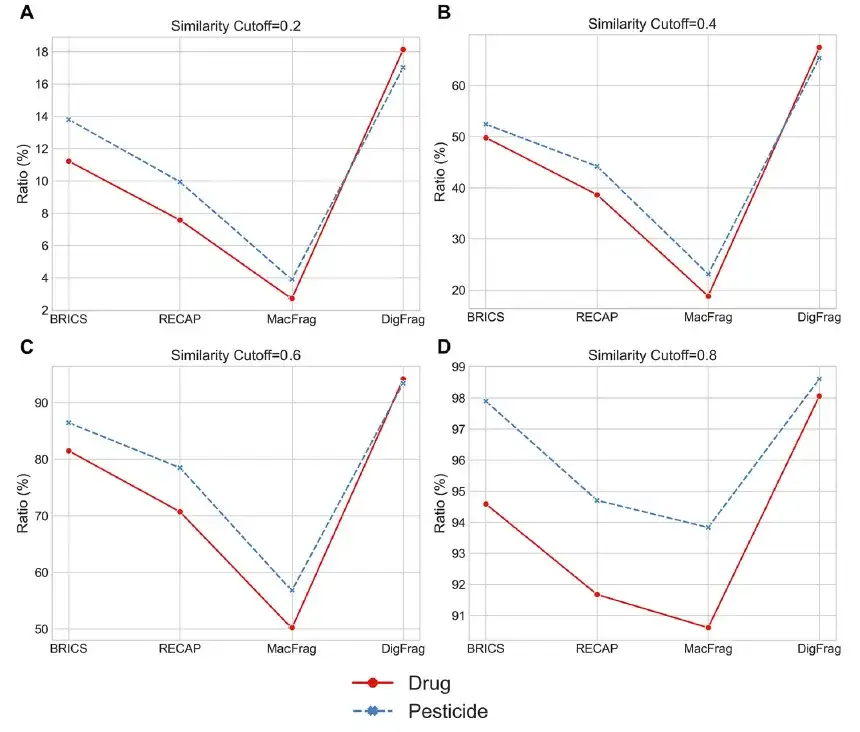
Cette étude s'est concentrée sur l'évaluation de la diversité structurelle des fragments segmentés tout en comparant la méthode DigFrag avec les méthodes traditionnelles (RECAP et BRICS) et une méthode de pointe (MacFrag). Les résultats ont montré que les fragments segmentés par DigFrag dans les médicaments et les pesticides avaient un taux de répétition inférieur à celui des trois autres méthodes, qui étaient respectivement 9,97%-21,37% et 8,94%-15,20%, indiquant qu'il peut générer des fragments uniques. MacFrag couvre la plupart des fragments de BRICS et RECAP, suggérant qu'il ne s'agit pas d'une innovation complète mais d'une extension des approches traditionnelles.

Les chercheurs ont également utilisé l’algorithme t-SNE pour visualiser la distribution de l’espace chimique. Comme le montre la figure ci-dessous, DigFrag fonctionne bien en termes de taux de regroupement de fragments, en particulier lorsque les seuils de similarité sont de 0,4 et 0,6, ce qui montre une diversité structurelle plus élevée.
Les modèles basés sur DigFrag génèrent des molécules de meilleure qualité
Sur la plateforme de référence MOSES, cette étude a comparé les performances de différents modèles génératifs. Comme le montrent les deux tableaux ci-dessous, le modèle basé sur DigFrag a obtenu un score de filtres de 0,828, montrant une sécurité plus élevée, qui peut être attribuée à la prise en compte complète de la toxicité et de la stabilité dans le processus de fragmentation de l'apprentissage en profondeur.
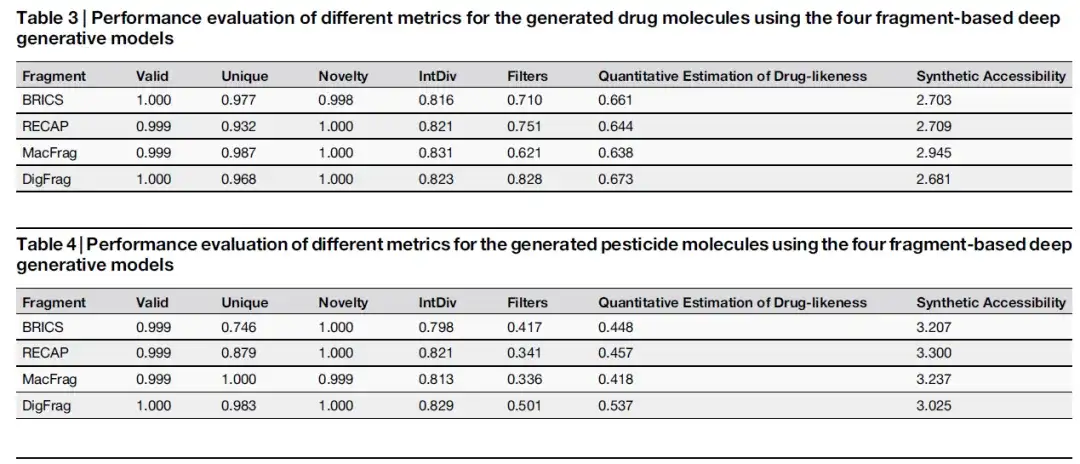
Comme le montre la figure ci-dessous, en termes de molécules de pesticides, les fragments moléculaires générés par le modèle basé sur DigFrag ont obtenu de bons résultats en termes de validité SMILES, de nouveauté, de diversité du squelette et d'alertes de structure. De plus, les fragments moléculaires de médicaments et de pesticides générés par le modèle DigFrag ont surpassé les autres modèles dans l’analyse de la valeur moyenne de l’estimation quantitative (QED) et l’accessibilité synthétique (SA).
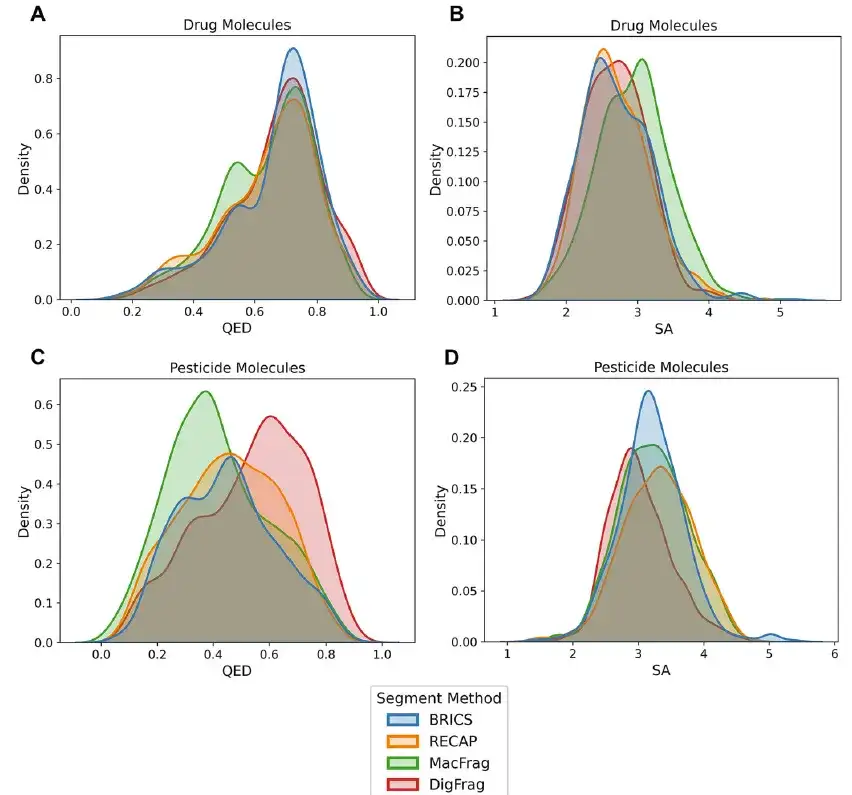
De plus, les fragments moléculaires segmentés par DigFrag présentent la plus grande similitude avec l'ensemble de données MOSES en termes de poids moléculaire, de QED et de distribution des propriétés SA. Ces résultats indiquent que le modèle DigFrag peut produire des molécules de meilleure qualité, tout en soulignant la préférence du modèle d'IA pour les données dérivées de l'IA dans la conception moléculaire, soulignant les avantages d'application de la technologie de l'IA dans ce domaine.
Sélection de 44 molécules de médicaments et de pesticides à haute efficacité et à faible énergie
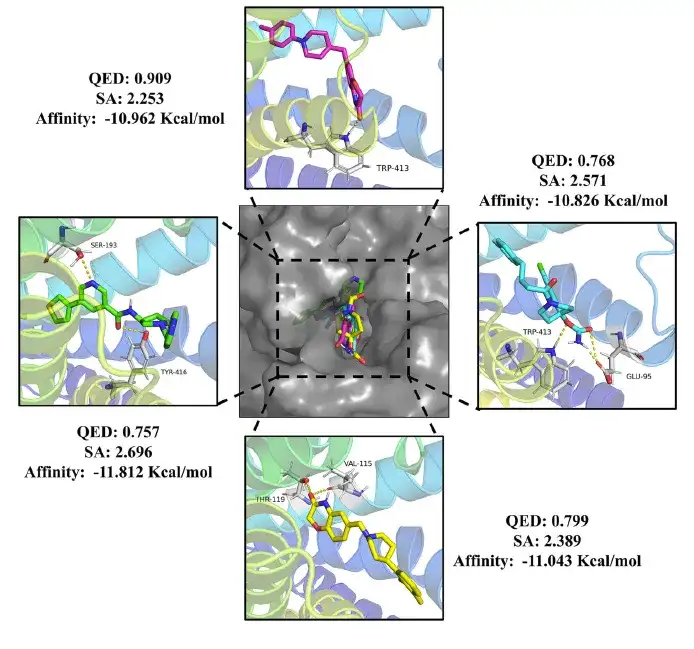
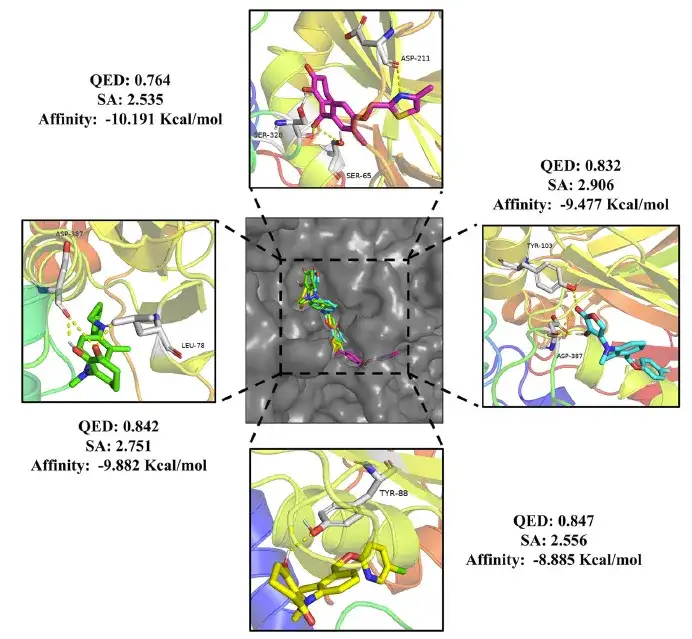
Finalement, après un criblage précis, l'étude a identifié 24 molécules médicamenteuses et 20 molécules pesticides, qui répondaient toutes aux critères de valeurs QED supérieures à 0,75, de valeurs SA inférieures à 3 et d'énergie libre de liaison inférieure à la dompéridone (-10,7 Kcal/mol) et à la méthotriazine (-8,4 Kcal/mol).
L’étude a également analysé les interactions de ces molécules avec leurs cibles. Comme le montre la figure ci-dessous, l’étude a révélé que la molécule de médicament peut se lier efficacement à la poche active DRD2 et former des liaisons hydrogène avec des résidus d’acides aminés clés.
De plus, comme le montre la figure ci-dessous, les molécules de pesticides sont liées de manière stable aux résidus d’acides aminés de l’HPPD en formant des liaisons hydrogène. Comparés aux médicaments positifs, les composés générés ont également présenté des modes de liaison différents, suggérant la possibilité de mécanismes pharmacologiques différents, ce qui ouvre de nouvelles perspectives pour les recherches futures.
L’application de l’IA dans la recherche sur les médicaments remodèle les règles du jeu
À ce stade, l’application de l’IA dans la recherche sur les médicaments devient de plus en plus approfondie. Grâce aux réseaux d’apprentissage profond, les modèles d’IA sont capables d’analyser des données biologiques et des structures chimiques complexes pour prédire l’activité et la sélectivité des molécules médicamenteuses.
L'équipe du professeur Yang Guangfu et du professeur associé Wang Fan mentionnée dans cette étude a également développé conjointement un modèle d'architecture d'apprentissage profond multimodal Pesti-DGI-Net pour prédire les propriétés de type pesticide plus tôt cette année. Il peut prédire les propriétés de type pesticide des composés en intégrant trois formes de représentation moléculaire : les descripteurs moléculaires, les images moléculaires et les graphiques moléculaires. Les résultats montrent que Pesti-DGI-Net présente des performances supérieures dans plusieurs indicateurs.
Lien vers l'article :
https://doi.org/10.1016/j.compag.2024.108660
En outre, l’IA a récemment obtenu des résultats fructueux dans le domaine de la recherche sur les médicaments. Il n'y a pas longtemps, l'Institut de nutrition et de santé de Shanghai de l'Académie chinoise des sciences a construit un modèle d'apprentissage profond à double vue JointSyn pour prédire les effets synergétiques des combinaisons de médicaments. Les résultats montrent que JointSyn surpasse les méthodes de pointe existantes en termes de précision de prédiction et de robustesse sur divers benchmarks.
Lien vers l'article :
https://doi.org/10.1093/bioinformatics/btae604
Outre son application dans la prédiction des propriétés des médicaments, la technologie de l’IA a également obtenu des résultats de recherche remarquables dans de nombreux domaines tels que l’optimisation de la conception des médicaments, l’évaluation de la toxicologie et de la sécurité, la conception des essais cliniques et la sélection des patients. On peut prévoir que l’application de l’IA dans la recherche sur les médicaments va remodeler les règles du jeu dans le développement des médicaments. Grâce aux progrès continus de la technologie, des options de traitement plus sûres et plus efficaces peuvent être proposées aux patients en améliorant la précision des prévisions, en optimisant la conception des médicaments et en réduisant les coûts et le temps de développement.








