Command Palette
Search for a command to run...
Réalisez La Prédiction De l'arrimage Dynamique Des Protéines ! L'Université Jiaotong De Shanghai, La Technologie Xingyao, l'Université Sun Yat-sen Et d'autres Ont Lancé Conjointement Le Modèle De Génération Géométrique Profonde DynamicBind

Les protéines sont la base matérielle de la vie. Sa fonction est étroitement liée à la dynamique de la structure et de la conformation des protéines et est régulée par des ligands. L’étude des interactions protéine-ligand est d’une grande importance pour la découverte et le criblage de médicaments. En revenant sur les progrès de ses recherches, le lancement d’AlphaFold constitue une avancée majeure qui permet de prédire la structure tridimensionnelle spatiale d’une seule protéine et de fournir une base structurelle pour l’étude des interactions protéine-ligand.
Cependant, AlphaFold ne peut prédire que la structure statique d'une protéine à un moment donné et ne parvient pas à prédire les changements dynamiques dans la structure de la protéine.Lorsque la structure protéique sans ligand prédite par AlphaFold est utilisée comme entrée pour l'amarrage, les prédictions de position du ligand résultantes ne correspondent souvent pas à la structure cocristalline liée au ligand. De plus, il est peu probable que la structure prédite par AlphaFold présente les configurations de chaîne latérale et de chaîne principale les plus favorables pour la liaison du ligand, ce qui fait que les sites actifs pertinents ne sont pas dans la bonne position. Il est donc actuellement difficile d’utiliser la structure AlphaFold pour le criblage et la conception de médicaments.
Compte tenu de cela,Le groupe de recherche dirigé par Zheng Shuangjia de l'Université Jiao Tong de Shanghai, en collaboration avec Star Pharma Technology, l'École de pharmacie de l'Université Sun Yat-sen et l'Université Rice, a proposé un modèle génératif géométrique profond DynamicBind conçu pour l'amarrage dynamique des protéines.Il peut ajuster efficacement la conformation des protéines de l'état initial prédit par AlphaFold à un état de type holo, fournissant un nouveau paradigme de recherche basé sur l'apprentissage en profondeur et prenant en compte les changements dynamiques des protéines pour le développement de médicaments à l'ère post-AlphaFold.Cette méthode a également été vérifiée par des expériences humides dans le cadre du concours international de criblage de médicaments CACHE, et permet de cribler des composés principaux compétitifs pour des cibles difficiles à cibler pour le traitement de la maladie de Parkinson.
L'étude, intitulée « DynamicBind : prédire la structure du complexe protéine-ligand spécifique au ligand avec un modèle génératif équivariant profond », a été publiée dans Nature Communications.
Points saillants de la recherche :
* En utilisant des modèles avancés de diffusion profonde et une technologie de réseau neuronal à géométrie équivariante, la génération de conformation des protéines et la prédiction de la pose des ligands sont unifiées dans un seul cadre, réalisant une prédiction d'arrimage dynamique des protéines et des ligands.
* DynamicBind surpasse les méthodes d'amarrage traditionnelles et les méthodes d'amarrage rigides basées sur l'apprentissage profond dans l'amarrage protéine-ligand
* DynamicBind utilise la conformation protéique prédite par AlphaFold pour ajuster dynamiquement la conformation protéique et trouver la conformation optimale qui convient le mieux au ligand

Adresse du document :
https://www.nature.com/articles/s41467-024-45461-2
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : basé sur l'ensemble de données PDBbind, l'ensemble de tests MDT est utilisé pour étendre la portée de l'évaluation
Les chercheurs ont d'abord utilisé l'ensemble de données PDBbind pour former les ensembles d'entraînement, de validation et de test du modèle par ordre chronologique, associés à des affinités de liaison mesurées expérimentalement.Étant donné que l'ensemble de tests PDBbind contient environ 300 structures de 2019, dont de nombreux ligands non à petites molécules (53 sont des peptides), les chercheurs ont élargi la portée de l'évaluation avec un ensemble de tests de cibles médicamenteuses majeures (MDT) organisé.
L'ensemble de tests MDT comprend 599 structures archivées en 2020 ou après, y compris des ligands de type médicament et des protéines de quatre grandes familles : kinases, GPCR, récepteurs nucléaires et canaux ioniques. Ces familles de protéines représentent les cibles d'environ 701 médicaments à petites molécules approuvés par TP3T et approuvés par la FDA, ce qui est représentatif.
DynamicBind : un modèle géométrique basé sur l'apprentissage profond pour prédire la structure des complexes dynamiques
Contrairement aux méthodes d'amarrage traditionnelles qui traitent les protéines comme des entités essentiellement rigides, DynamicBind utilise des modèles avancés de diffusion profonde et une technologie de réseau neuronal à géométrie équivariante pour unifier les deux étapes traditionnellement distinctes de la génération de conformation des protéines et de la prédiction de la pose des ligands dans un cadre unique, permettant ainsi une prédiction d'amarrage dynamique des protéines et des ligands.En même temps, en tant que méthode d’apprentissage profond de bout en bout, elle est également plusieurs ordres de grandeur plus rapide que les simulations MD traditionnelles pour échantillonner une large gamme de changements conformationnels de protéines.
DynamicBind accepte les structures de type apo au format PDB et plusieurs formats de ligands de petites molécules largement utilisés, tels que SMILES ou SDF.Lors de l'inférence, le modèle place aléatoirement des ligands et des conformations de départ de ligands sont générées autour de la protéine à l'aide de RDKit. Au cours de la phase de formation, le modèle vise à apprendre le processus de la conformation de type apo à la conformation holo. Lors de l’inférence, le modèle itère 20 fois sur la structure d’entrée initiale.
Comme le montre la figure a ci-dessous, le rose représente l'état holographique (holo) de la conformation de la protéine, le vert représente l'apolipoprotéine initiale et les conformations prédites par le modèle, le cyan représente le ligand natif et l'orange représente le ligand prédit.
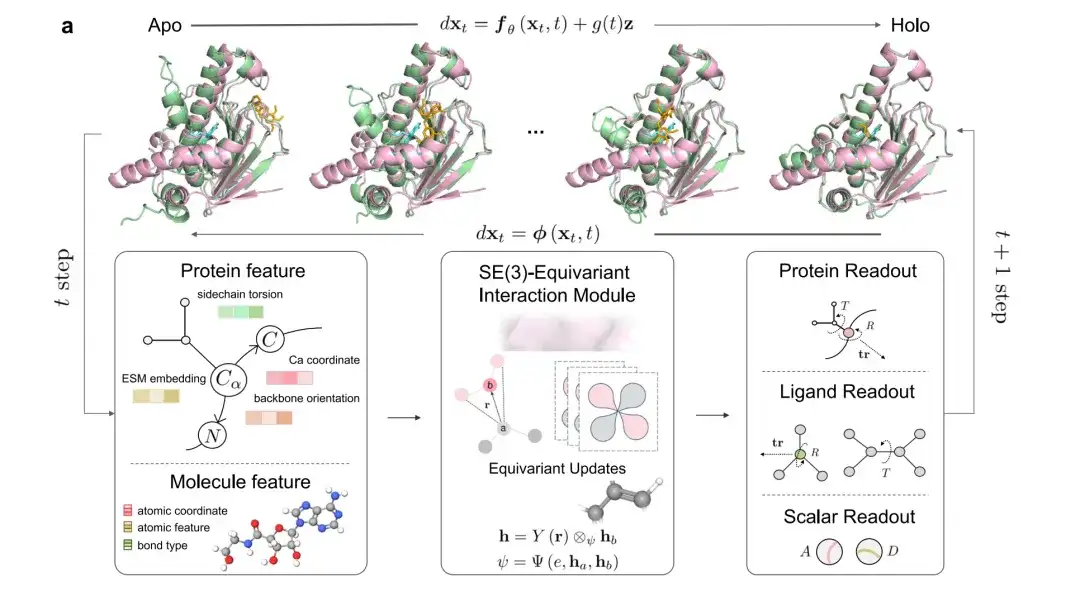
À chaque itération, les caractéristiques et coordonnées de la protéine et du ligand (y compris la torsion de la chaîne latérale, les coordonnées atomiques du Ca, etc.) sont entrées dans un module d'interaction équivariante SE(3). Les résultats de sortie du modèle incluent la translation et la rotation globales du ligand et de chaque résidu protéique, l'angle de torsion du ligand et la rotation de l'angle chi des résidus protéiques, ainsi que deux modules de prédiction (affinité de liaison A et score de confiance D).
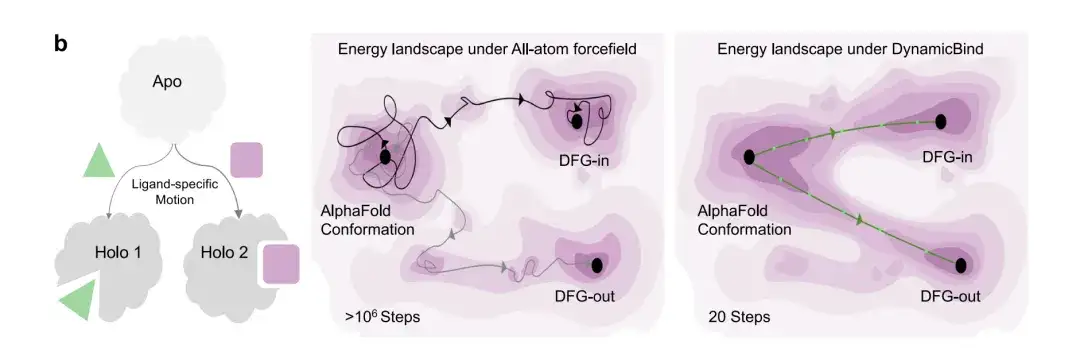
La figure b ci-dessous montre l’efficacité d’échantillonnage du modèle DynamicBind. Au cours de la transition DFG-in vers DFG-out de la protéine kinase, le modèle peut prédire deux conformations holo différentes lorsque la protéine se lie à deux ligands différents. De plus, DynamicBind peut prédire la conformation de la protéine liée en 20 étapes, tandis que les simulations MD tout atome nécessitent des millions d'étapes pour trouver le même état de liaison.
DynamicBind est un outil polyvalent dans la prédiction de l'arrimage dynamique des protéines. Il fonctionne bien dans cinq tâches principales.
Pour évaluer les performances du modèle de DynamicBind, les chercheurs l'ont testé sur cinq tâches, notamment :
(1) Comparez DynamicBind aux méthodes d'amarrage actuelles ;
(2) la capacité d’échantillonner les changements conformationnels dans un grand nombre de protéines ;
(3) la portée de l’étude des changements conformationnels des protéines ;
(4) la capacité de prédire les poches cachées pour réaliser un amarrage dynamique ;
(5) Performance de dépistage dans les tests de référence sur les antibiotiques.
DynamicBind surpasse les méthodes d'ancrage traditionnelles et les méthodes d'ancrage rigides basées sur l'apprentissage profond
Lors des tests, les chercheurs n'ont pas utilisé de structures holographiques comme entrée et ont supposé que les conformations protéiques holographiques n'étaient pas disponibles, en utilisant uniquement les conformations protéiques prédites par AlphaFold comme entrée.Étant donné que les conformations holo présentent une forte complémentarité de forme et de charge avec les ligands cocristallisés, le processus de prédiction de la pose du ligand est simplifié.
Comme le montrent les figures a et b ci-dessous, les chercheurs ont comparé DynamicBind avec d’autres modèles de base sur l’ensemble de données PDBbind et l’ensemble de données MDT. Sous différents seuils RMSD, DynamicBind a surpassé les autres méthodes. Plus précisément, la proportion de ligands DynamicBind avec des seuils RMSD inférieurs à 2 Å (5 Å) est de 331 TP3T (651 TP3T) sur l'ensemble de tests PDBbind et de 391 TP3T (681 TP3T) sur l'ensemble de tests MDT.
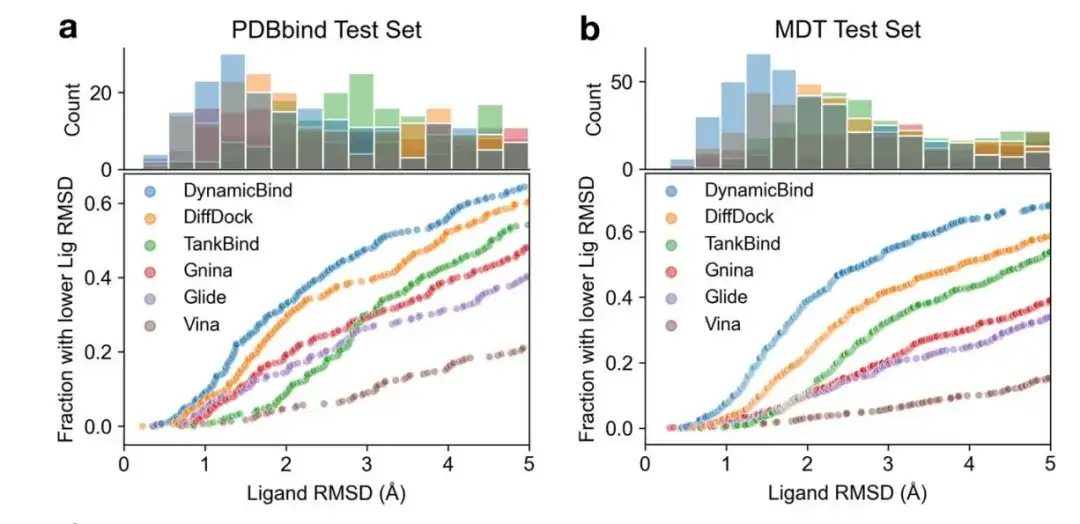
Cependant, lors de l'évaluation du modèle, seule la valeur RMSD du ligand est utilisée pour l'évaluation, ce qui est bénéfique pour les modèles basés sur l'apprentissage profond tels que DiffDock, TankBind et DynamicBind car ils ont une tolérance plus élevée aux conflits conformationnels ; mais elle est défavorable aux méthodes d'amarrage Gnina, Glide et Vina qui mettent en œuvre strictement les forces de Van der Waals et sont basées sur des champs de force, affectant ainsi l'objectivité de l'évaluation du modèle. Par conséquent, les chercheurs ont utilisé le RMSD du ligand et les scores de conflit pour évaluer le taux de réussite de la prédiction du ligand.
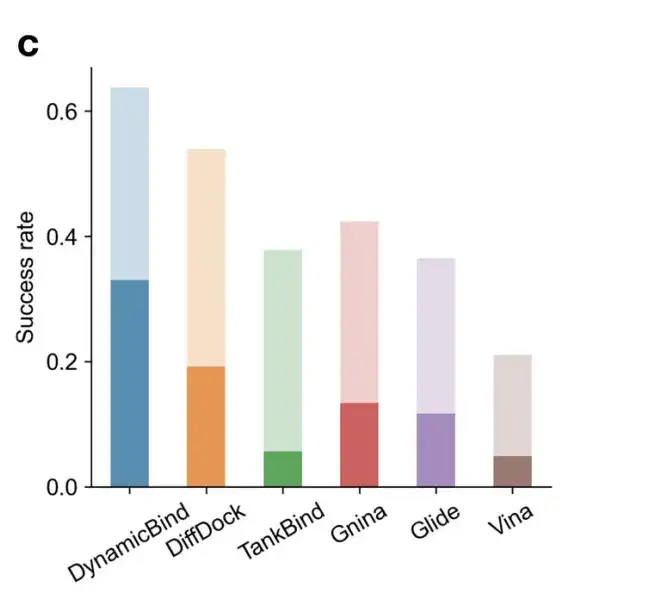
La figure c montre le taux de réussite de la prédiction du ligand en utilisant des critères stricts (ligand RMSD < 2 Å, score de conflit < 0,35) et des critères plus souples (ligand RMSD < 5 Å, score de conflit < 0,5). Dans des conditions plus strictes, le taux de réussite de DynamicBind (0,33) est 1,7 fois supérieur à celui du meilleur DiffDock de référence (0,19).
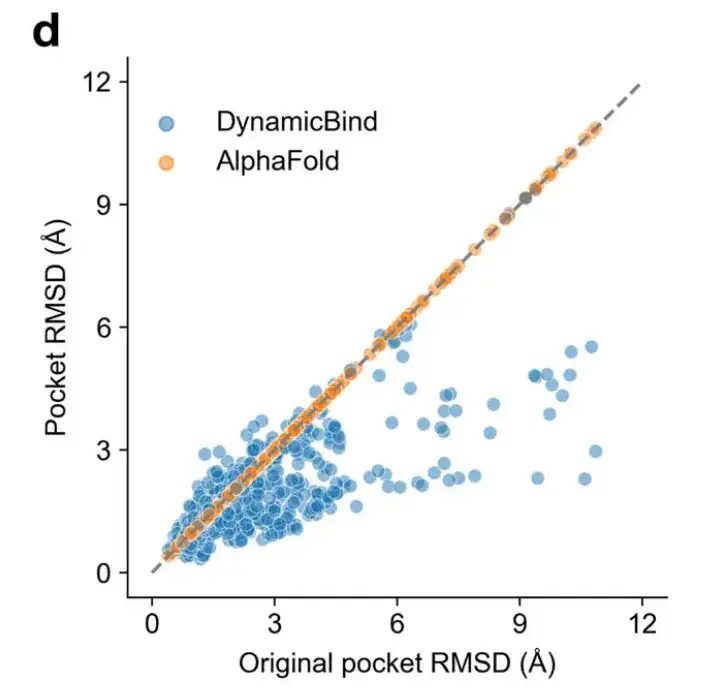
De plus, même lorsque le RMSD entre la poche initiale et la structure cristalline est grand, le RMSD de poche prédit par DynamicBind est significativement plus petit que celui prédit par AlphaFold, comme le montre la figure d ci-dessous.
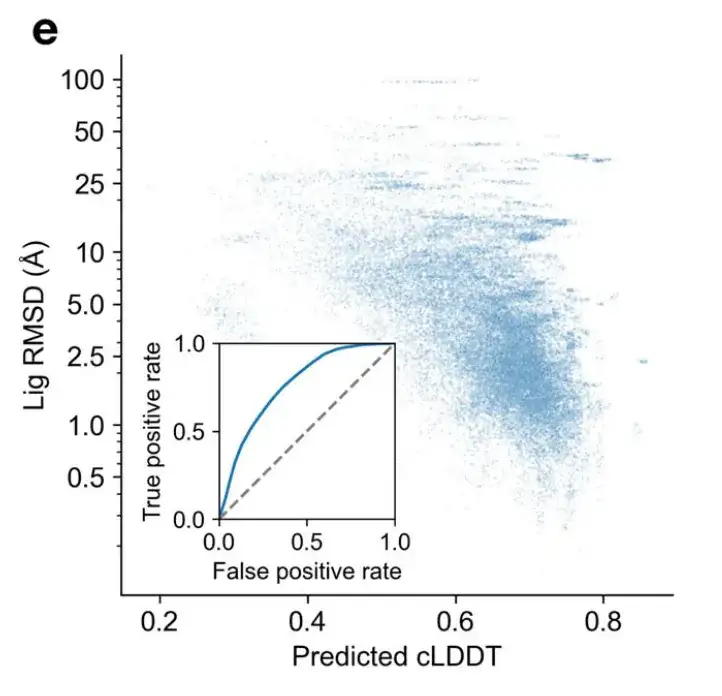
Compte tenu de la capacité de DynamicBind à générer diverses conformations et inspirés par les scores LDDT d'AlphaFold, les chercheurs ont développé un module de notation par contact-LDDT (cLDDT) pour sélectionner la structure complexe la plus appropriée à partir de la sortie prédite.
Comme le montre la figure e ci-dessous, le cLDDT prédit par DynamicBind présente une bonne corrélation avec le RMSD réel du ligand, indiquant son efficacité dans la sélection de structures complexes de haute qualité.
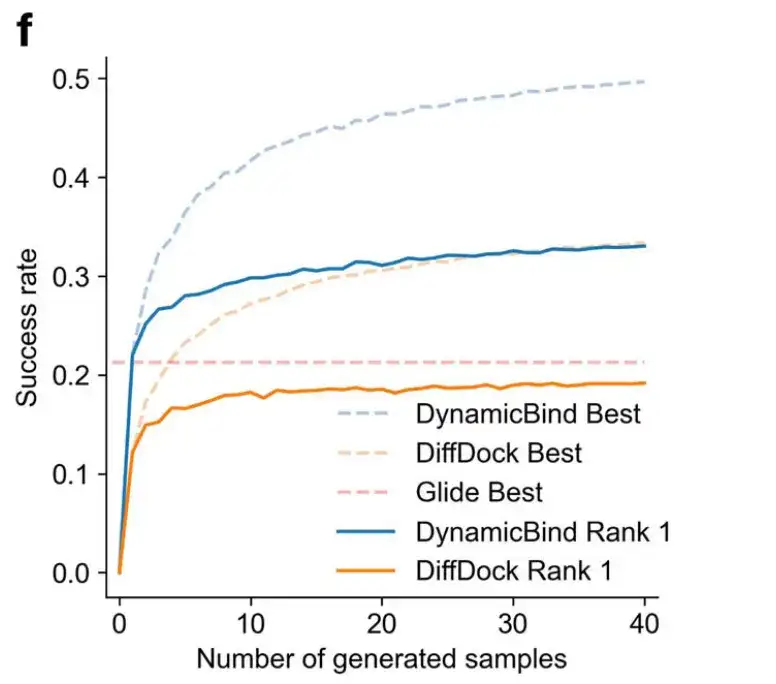
Comme le montre la figure f ci-dessous, à mesure que le nombre d’échantillons générés augmente, le taux de réussite du modèle DynamicBind dans la prédiction des poses de ligands augmente également.
DynamicBind peut capturer les changements conformationnels des protéines spécifiques au ligand
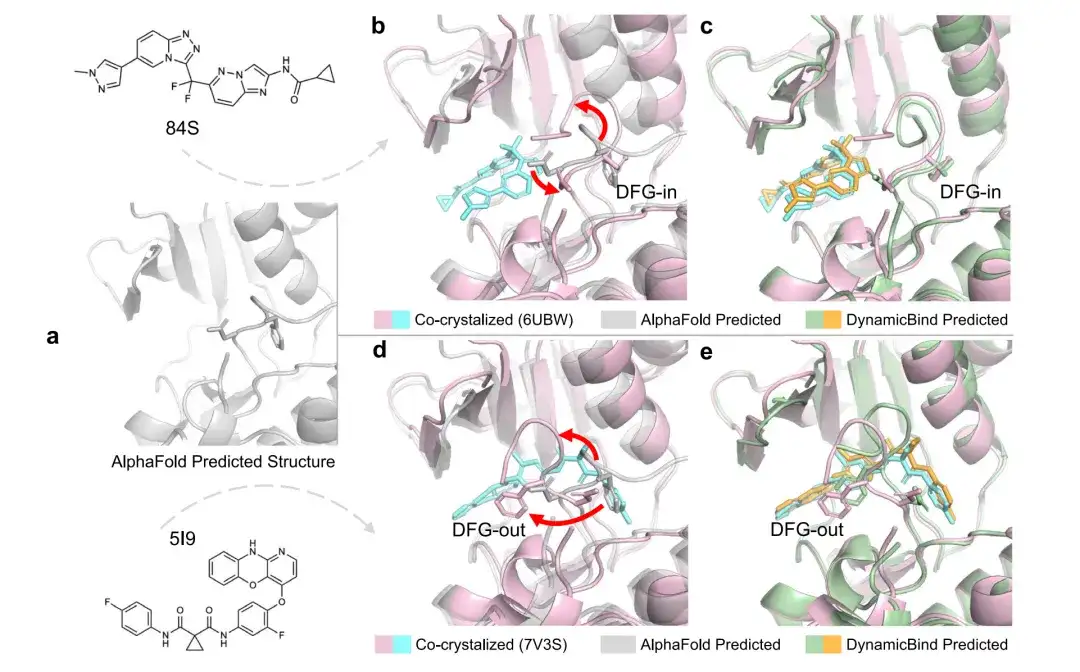
Les protocoles d’amarrage traditionnels effectuent généralement l’échantillonnage de la conformation des protéines dans le cadre d’une étape distincte du processus d’amarrage. Cependant, dans de nombreux cas, deux ligands différents peuvent s’intégrer dans des conformations protéiques mutuellement exclusives. Dans les modèles d'amarrage précédents, la protéine devait être préréglée dans la conformation correcte avant qu'il soit possible d'identifier la posture de liaison appropriée du ligand.En revanche, DynamicBind utilise la conformation protéique prédite par AlphaFold pour ajuster dynamiquement la conformation protéique et trouver la conformation optimale qui convient le mieux au ligand d'intérêt, voir la figure a ci-dessous.
Les figures b à e montrent le RMSD des ligands et des poches prédits par DynamicBind et AlphaFold dans les structures PDB 6UBW et PDB 7V3S. Pour PDB 6UBW, DynamicBind prédit un RMSD de ligand de 0,49 Å et un RMSD de poche de 1,97 Å, tandis que le RMSD de poche de la structure AlphaFold est de 9,44 Å. Pour PDB 7V3S, DynamicBind prédit un RMSD de ligand de 0,51 Å et un RMSD de poche de 1,19 Å (AlphaFold 6,02 Å).
Les figures f et g montrent comment les protéines marquées par UniProt ID, à partir de la même structure initiale, se déplacent progressivement vers la conformation DFG-in après la liaison aux inhibiteurs de type I et tendent vers la conformation DFG-out lors de l'interaction avec les inhibiteurs de type II.
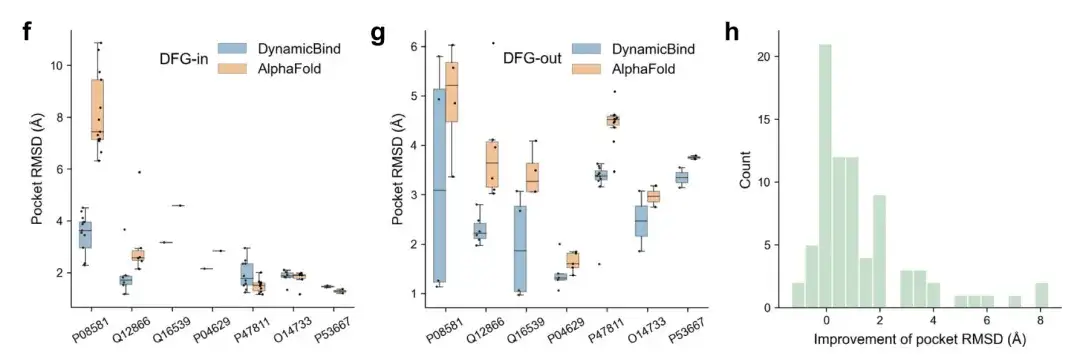
La figure h révèle que la plupart des structures protéiques prédites par DynamicBind présentent un RMSD de poche inférieur à celui de la structure AlphaFold initiale.
Les résultats ci-dessus démontrent que DynamicBind est capable de capturer les changements conformationnels spécifiques au ligand.Autrement dit, DynamicBind peut identifier les composés qui se lient bien à d’autres conformations possibles de la protéine, même si la conformation particulière diffère de la structure protéique initialement fournie.
DynamicBind couvre plusieurs échelles de changements conformationnels des protéines
Les chercheurs ont évalué DynamicBind en utilisant six types différents de changements conformationnels à plusieurs échelles, allant des picosecondes aux millisecondes.Comme le montre la figure ci-dessous, le rose représente la structure cristalline, le blanc représente la structure AlphaFold, le vert représente la structure prédite par DynamicBind, le cyan représente le ligand natif et l'orange représente le ligand prédit par DynamicBind.
Sur la base d'une comparaison avec la structure cristalline,Δpocket RMSD mesure la différence de RMSD de poche entre la structure protéique prédite par le modèle et la structure AlphaFold.Un RMSD Δpocket négatif indique que la structure prédite par DynamicBind est plus proche de la structure prédite par AlphaFold que de la structure cristalline.
Δclash mesure la différence des scores de conflit entre la paire protéine-ligand prédite et le ligand greffé dans la structure AlphaFold.Un Δclash négatif indique moins de conflits dans le complexe prédit.
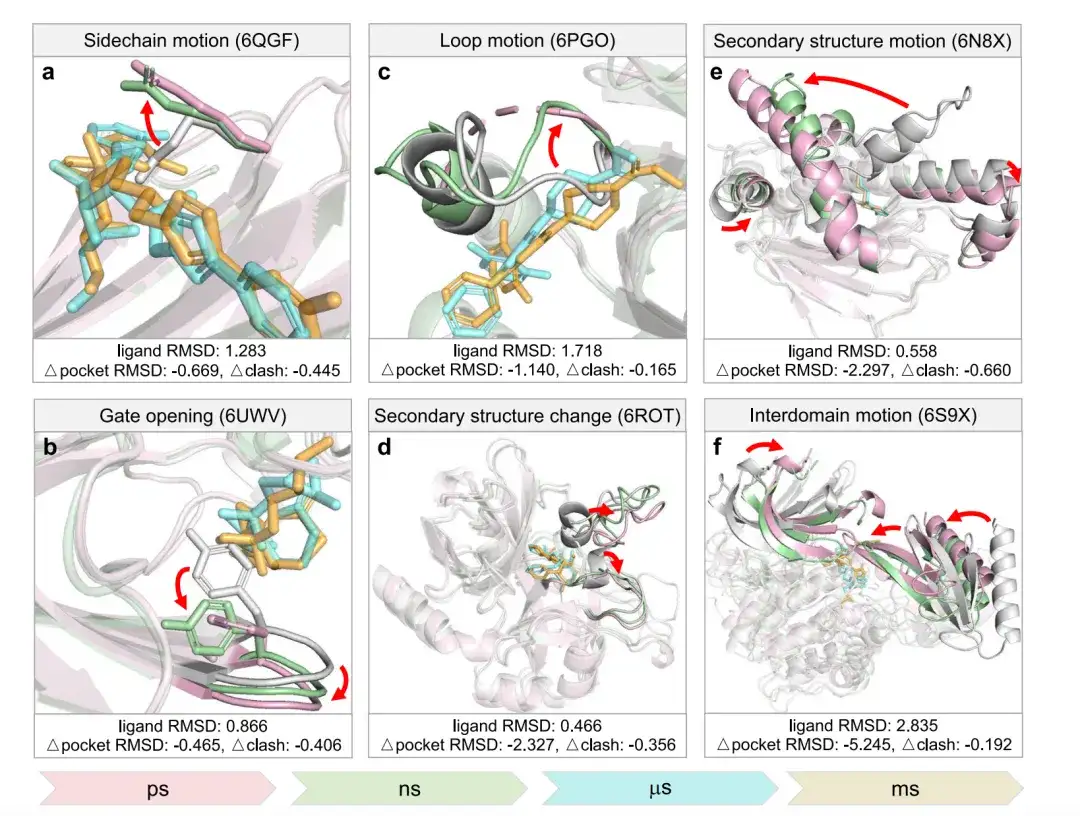
Dans la figure a, le ligand natif entre en conflit avec une chaîne latérale de la structure AlphaFold superposée ; dans la prédiction DynamicBind, cette chaîne latérale se déplace vers la conformation native, résolvant ainsi le conflit. Dans la figure b, une tyrosine dans la structure AlphaFold bloque une partie de la poche ; dans la structure prédite DynamicBind et la structure native, cette partie de la poche devient accessible. Dans le panneau c, une boucle flexible croise le ligand, alors qu'elle est éloignée dans la prédiction DynamicBind, ce qui est cohérent avec la structure native.
Dans la figure d, l’hélice α devient une boucle près du site de liaison du ligand. Dans la figure e, la structure secondaire de la protéine de choc thermique Hsp90α subit un grand mouvement lorsqu'elle passe d'un état fermé à un état ouvert. Dans le panneau f, les deux domaines de la kinase AKT1 se condensent ensemble pour former une poche qui n'existait pas auparavant.
En résumé, lorsque la poche de liaison du ligand n'est pas suffisamment spacieuse ou ne forme pas la conformation prédite par AlphaFold, le modèle DynamicBind peut prédire divers changements conformationnels associés à la liaison du ligand.
DynamicBind identifie les sites de liaison cryptiques
Les protéines génèrent souvent des poches cryptiques au cours de processus dynamiques, ce qui peut révéler des sites médicamenteux qui ne sont pas découverts dans les structures statiques, faisant ainsi des protéines auparavant « non médicamenteuses » des cibles médicamenteuses potentielles.En utilisant la protéine contenant le domaine SET 2 (SEtD2) comme étude de cas, les chercheurs ont démontré l'utilité de DynamicBind pour révéler ces poches cryptiques.
SETD2, une histone méthyltransférase qui est un médicament clé pour le traitement du myélome multiple (MM) et du lymphome diffus à grandes cellules B (DLBCL), possède une poche cryptique et est la cible d'un composé hautement sélectif, EZM0414, actuellement en phase I d'essais cliniques.
Comme le montrent les figures a et b ci-dessous, tous les homologues de SETD2 dans l'ensemble d'entraînement (défini par une similarité protéique Smith-Waterman de plus de 0,4) ont co-cristallisé avec la S-adénosylméthionine (SAM) ou les analogues de la sinefungine, représentés par des lignes. La barre cyan indique le ligand EZM0414 de PDB 7TY2 et la barre rose indique la protéine.
Dans la figure c, le blanc représente la structure AlphaFold et sa surface, où les sites cryptiques sont bloqués, ce qui entraîne un grand nombre de conflits avec l'EZM0414 transplanté.
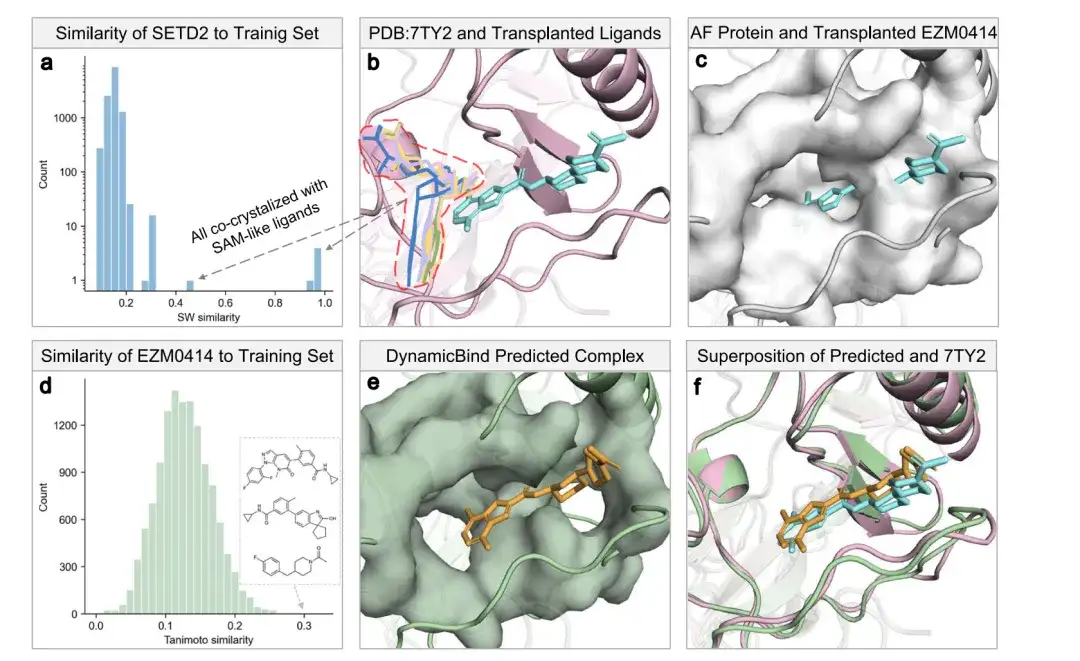
Le panneau d confirme que l'EZM0414 est un ligand invisible et que même le ligand Tanimoto le plus similaire s'écarte considérablement de l'EZM0414. La figure e montre la structure du complexe protéine-ligand prédite par le modèle DynamicBind, qui prend comme entrée la structure SETD2 prédite par AlphaFold et la représentation SMILES de EZM0414. La figure f montre le chevauchement entre la structure du complexe protéine-ligand prédite par DynamicBind et la structure cristalline du complexe SETD2-EZM0414 (PDB 7TY2).
À en juger par les résultats, DynamicBind a réussi à arrimer dynamiquement la poche cachée, non seulement en plaçant avec succès le ligand, mais également en trouvant une conformation de poche plus appropriée (le RMSD du ligand obtenu était de 1,4 Å et le RMSD de la poche était de 2,16 Å).
DynamicBind obtient de meilleures performances de criblage de médicaments sur les critères de référence des antibiotiques
Dans le processus de découverte de médicaments ciblés, le criblage de candidats médicaments potentiels et le contre-criblage (identification de cibles protéiques pour des composés spécifiques) sont essentiels.Pour évaluer les performances de criblage du modèle DynamicBind dans la pratique, les chercheurs ont ajouté un module de prédiction d'affinité au modèle, l'ont formé à l'aide de données d'affinité de liaison mesurées expérimentalement obtenues à partir de l'ensemble de données PDBbind et l'ont évalué sur des données de test de criblage de médicaments provenant du protéome antibiotique publié en 2023 (y compris 12 cibles protéiques et près de 3 000 données d'activité mesurées).
Comme le montre la figure a ci-dessous, DynamicBind surpasse les méthodes d'amarrage courantes telles que VINA et DOCK6.9, ainsi que la meilleure méthode de re-scoring basée sur l'apprentissage automatique, avec une zone moyenne sous la courbe caractéristique de fonctionnement du récepteur (auROC) de 0,68. Cette amélioration des performances est due à la capacité d'ancrage dynamique de DynamicBind, qui peut affiner la structure AlphaFold vers un état natif plus proche, permettant ainsi une estimation plus précise de l'affinité de liaison.
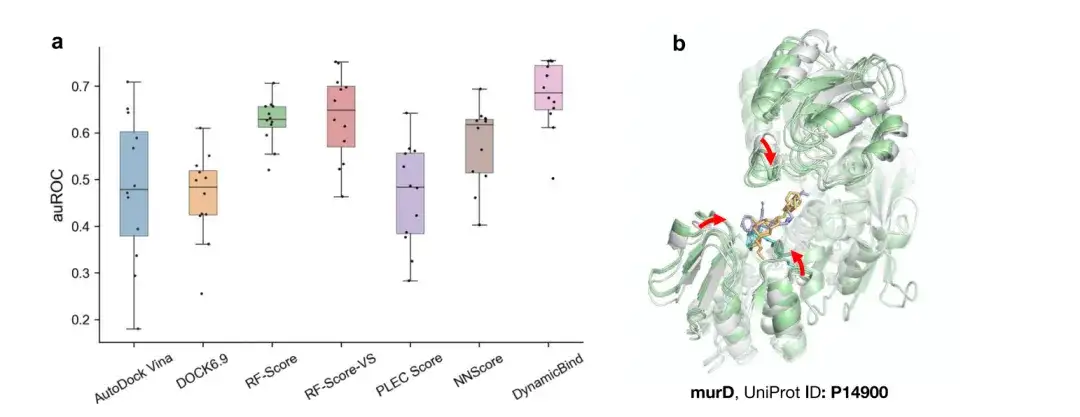
La figure b ci-dessus montre que la structure protéique murD prédite par DynamicBind entoure le ligand plus étroitement, formant davantage d'interactions qui étaient impossibles à former dans la structure initiale d'AlphaFold.
Ces résultats indiquent que DynamicBind surpasse systématiquement les méthodes d'amarrage traditionnelles et les méthodes d'amarrage rigide basées sur l'apprentissage profond, et le modèle présente un grand potentiel dans les applications de criblage virtuel au niveau du protéome en raison de ses capacités de prédiction d'affinité de liaison.
Décoder la structure et la fonction complexes des protéines pour contribuer à la découverte de médicaments intelligents
Basé sur la prédiction de structure statique d'AlphaFold, le modèle DynamicBind introduit de manière innovante la technologie d'intelligence artificielle générative, résolvant avec succès le défi de la prédiction de structure complexe dynamique. Prédire les changements dynamiques dans la structure des protéines est d’une grande importance pour comprendre les processus vitaux et développer de nouveaux médicaments. En particulier dans le développement de médicaments par IA, cela peut considérablement améliorer la précision et l’efficacité clinique du dépistage des médicaments par IA.
En tant que l'un des principaux participants à ce résultat de recherche, le groupe de recherche de Zheng Shuangjia est depuis longtemps profondément engagé dans la recherche transversale de pointe sur l'intelligence artificielle générative et le développement de médicaments, et a obtenu des résultats fructueux.
Le 21 juin 2024, le groupe de recherche de Zheng Shuangjia a proposé une méthode d'apprentissage intermodal qui utilise des images de microscopie cellulaire à haut contenu perturbées au niveau phénotypique pour aider à l'apprentissage de la représentation moléculaire.Cette approche peut efficacement établir un pont entre les molécules et la caractérisation, ce qui est d’une grande importance pour le développement de médicaments. La recherche connexe a été publiée dans Advanced Science sous le titre « Cross-Modal Graph Contrastive Learning with Cellular Images ».
Adresse du document :
https://onlinelibrary.wiley.com/doi/10.1002/advs.202404845
Le 25 mai 2024, le groupe de recherche de Zheng Shuangjia a proposé le cadre d'apprentissage multi-échelle MUSE, qui intègre efficacement les informations multi-échelles entre les échelles de structure atomique et de réseau moléculaire.Démontre le potentiel d’étendre la découverte informatique de médicaments à d’autres échelles. La recherche connexe a été publiée dans Nature Communications sous le titre « Un cadre variationnel d’espérance-maximisation pour un apprentissage multi-échelle équilibré des interactions entre protéines et médicaments ».
Adresse du document :
https://www.nature.com/articles/s41467-024-48801-4
Le 15 septembre 2022, le groupe de recherche de Zheng Shuangjia a développé un algorithme de conception de médicaments intelligent génératif pour les cibles difficiles à traiter et a conçu les composés phares PROTAC dans un court laps de temps.Cela a été vérifié par des expériences sur des animaux, démontrant l’énorme potentiel de l’intégration des technologies de l’information et de la biotechnologie. Cette série de résultats a reçu des citations et des évaluations positives de la part des meilleurs groupes de recherche du domaine, notamment l’équipe Google DeepMind AlphaFold et l’équipe du professeur David Baker de l’Université de Washington. La recherche connexe a été publiée dans Nature Machine Intelligence sous le titre « Conception rationnelle accélérée de PROTAC via l'apprentissage en profondeur et les simulations moléculaires ».
Adresse du document :
https://www.nature.com/articles/s42256-022-00527-y
Le 14 février 2020, le groupe de recherche de Zheng Shuangjia a proposé un système de questions-réponses quasi visuel basé sur un cadre d'apprentissage profond de bout en bout.Pour identifier l’interaction entre les médicaments et les protéines, il facilite efficacement la découverte de médicaments. La recherche connexe a été publiée dans Nature Machine Intelligence sous le titre « Prédire l’interaction médicament-protéine à l’aide d’un système de questions-réponses quasi-visuel ».
Adresse du document :
https://www.nature.com/articles/s42256-020-0152-y
Sur la base de leur compréhension de la recherche de pointe transversale sur l'intelligence artificielle générative et le développement de médicaments, le groupe de recherche de Zheng Shuangjia se concentre sur la conception intelligente de médicaments pour les maladies liées au métabolisme et au vieillissement, crée un nouveau modèle de développement de médicaments qui intègre l'informatique et la BT, et s'engage à contribuer davantage à la découverte de médicaments intelligents de bout en bout.
Références :








