Command Palette
Search for a command to run...
Publié Dans La Revue Nature ! Le Premier Auteur De l'article Explique En Détail La Méthode d'apprentissage Par Petits Échantillons Du Modèle De Langage Protéique Pour Résoudre Le Problème Du Manque De Données Expérimentales humides.

Dans le troisième épisode de la série « Meet AI4S », nous avons l'honneur d'inviter Zhou Ziyi, chercheur postdoctoral à l'Institut des sciences naturelles de l'Université Jiao Tong de Shanghai et au Centre national de mathématiques appliquées de Shanghai,Son groupe de recherche à l'Université Jiao Tong de Shanghai, le groupe de Hong Liang, se concentre sur la conception de protéines et de médicaments IA et sur la biophysique moléculaire. L’équipe de recherche a obtenu des résultats fructueux. À ce jour, ils ont publié 77 articles de recherche, dont beaucoup ont été publiés dans des revues Nature.
Lors de cette séance de partage, le Dr Zhou Ziyi a partagé les derniers résultats de recherche de l'équipe sous le titre « Méthode d'apprentissage par petits échantillons pour le modèle de langage des protéines » et a exploré de nouvelles idées pour l'évolution dirigée assistée par l'IA.
Contexte de la recherche sur le modèle de langage des protéines (PLM)
Protéines et ingénierie des protéines
Les protéines sont le principal vecteur des fonctions biologiques et l’exécuteur des activités vitales. L'acide aminé naturel ammoniac subit une réaction de condensation par déshydratation pour former la séquence résiduelle de la protéine, qui est ensuite repliée dans une structure tertiaire. La modification du profil d’acides aminés d’une protéine peut affecter sa structure et sa fonction.
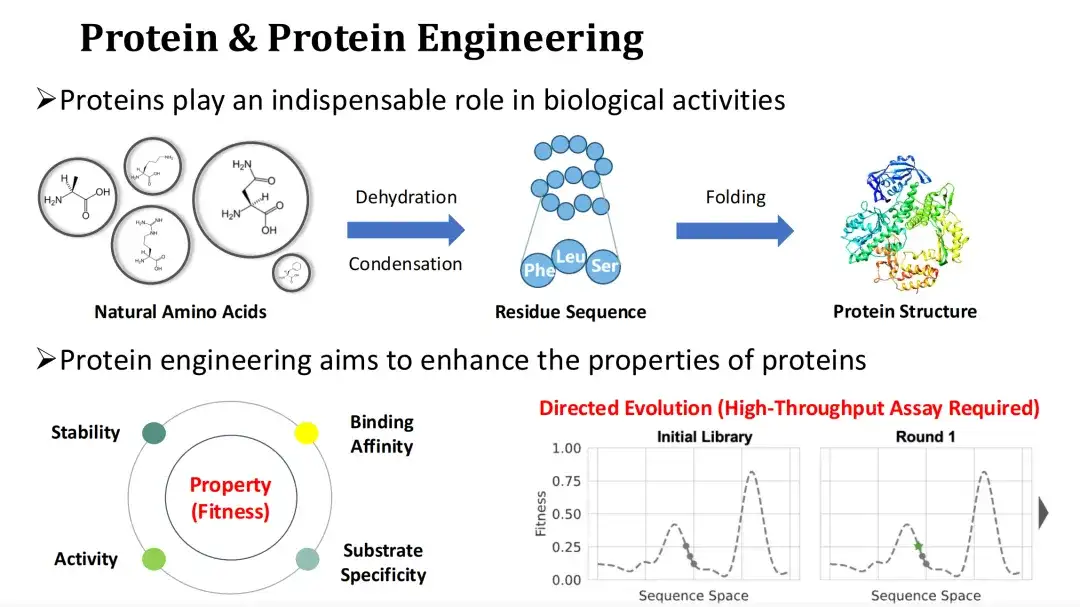
Étant donné que les protéines naturelles sont souvent difficiles à satisfaire aux besoins industriels ou médicaux, l’ingénierie des protéines espère améliorer les propriétés fonctionnelles des protéines, telles que l’activité catalytique, la stabilité, la capacité de liaison, etc., en les mutant.
Nous appelons généralement la quantification des propriétés fonctionnelles des protéines la forme physique. L’évolution dirigée est désormais la méthode d’ingénierie des protéines la plus répandue.Elle repose sur une mutagenèse aléatoire et des expériences à haut débit pour trouver des mutants dotés d’une bonne condition physique, mais les coûts expérimentaux sont élevés. Compte tenu de cela,Le sujet que je partagerai aujourd’hui est de savoir comment utiliser les méthodes d’IA pour prédire la condition physique et ainsi réduire les coûts expérimentaux.
Architecture PLM
Nous savons que les modèles de langage représentés par ChatGPT sont très puissants et capables de comprendre et de générer du texte de haute qualité. Ces modèles de langage sont pré-entraînés sur des quantités massives de texte et sont capables d'apprendre les lois statistiques du texte et de maîtriser la grammaire de base et la sémantique des mots en contexte. Alors, est-il possible d’entraîner des modèles de langage protéique de manière similaire sur des séquences protéiques massives ? La réponse est oui.

Le modèle de langage protéique PLM a trois fonctions principales. Premièrement, le PLM peut modéliser les informations co-évolutives des séquences protéiques et apprendre les interdépendances et les contraintes évolutives entre les résidus.Tout comme un langage naturel, LM peut apprendre la grammaire d’un texte. Le PLM peut utiliser cette capacité pour estimer quelles mutations sont nuisibles ou bénéfiques, et ainsi prédire l’adéquation de la mutation.
Deuxièmement, en plus de la prédiction de la condition physique, PLM peut également calculer la représentation vectorielle des protéines.Ces représentations peuvent être utilisées pour la prédiction de structure ou l'extraction de protéines et, après un réglage précis, peuvent également effectuer une prédiction de fonction.
Enfin, PLM peut effectuer une génération de protéines conditionnelle comme ChatGPT pour réaliser une conception de protéines de novo.
L'architecture du PLM est similaire à celle du langage naturel LM, qui est divisé en modèle autorégressif et modèle masqué.La structure du réseau de ces deux modèles utilise tous deux Transformer, qui se compose d'un mécanisme d'auto-attention et d'une couche entièrement connectée. La principale différence réside dans les objectifs de pré-formation.
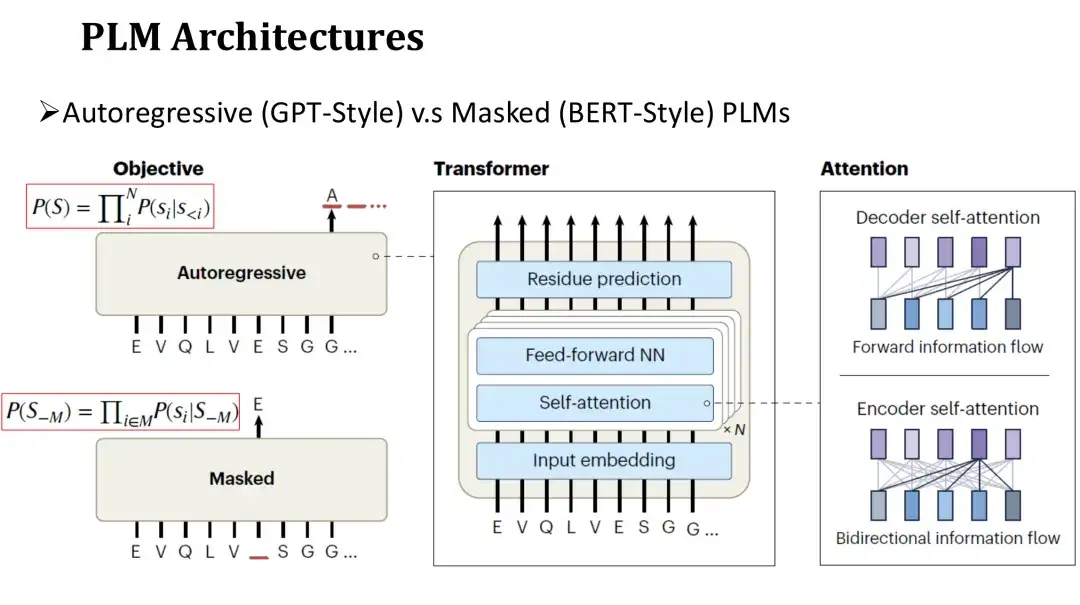
L’objectif de pré-formation du modèle autorégressif est de générer l’acide aminé suivant dans la séquence de gauche à droite.L’objectif du modèle de masquage est de restaurer les acides aminés masqués de manière aléatoire, de manière similaire au remplissage des blancs. Étant donné que le modèle autorégressif ne peut s’appuyer que sur la séquence générée à gauche pour prédire chaque acide aminé, son attention est unidirectionnelle.Le modèle de masquage peut voir les acides aminés des deux côtés de la position masquée pendant la prédiction.Son attention est donc à double sens.
Deux axes de recherche importants du PLM
À l’heure actuelle, les domaines de recherche du PLM sont principalement divisés en deux directions. Le premier est le PLM augmenté par la récupération.Lors de la formation ou de la prédiction, ce type de modèle prend l'alignement de séquences multiples (MSA) de la protéine actuelle comme entrée supplémentaire et améliore les performances de prédiction grâce aux informations récupérées. Par exemple, MSA Transformer et Tranception sont des modèles typiques de ce type.
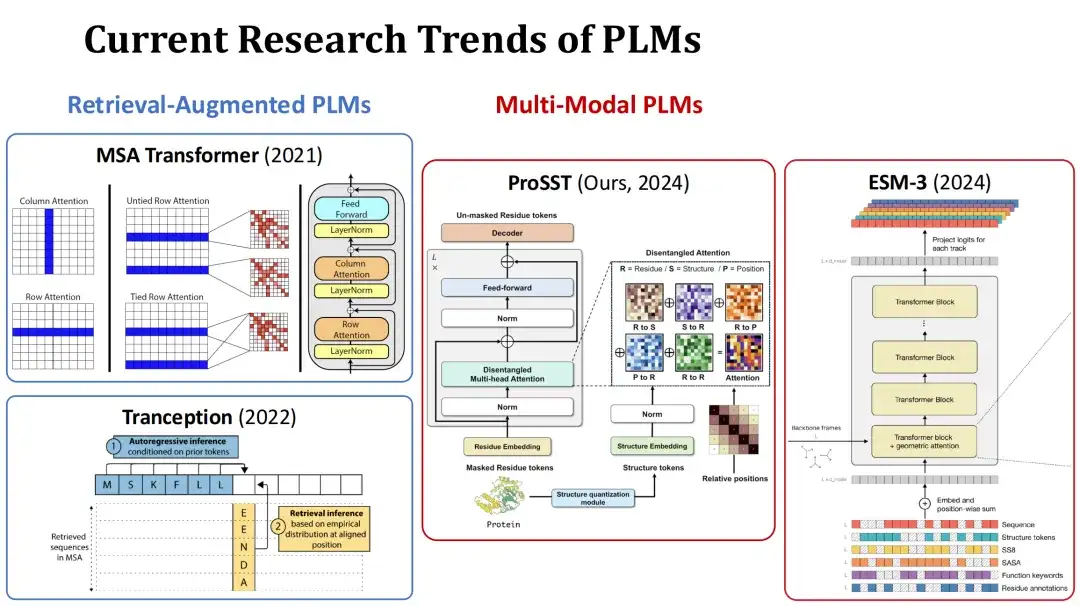
Le deuxième est le PLM multimodal.En plus des séquences protéiques, ce type de modèle prend également en compte la structure des protéines ou d'autres informations comme données d'entrée supplémentaires pour améliorer la capacité de représentation du modèle. Par exemple, le modèle ProSST soumis par notre groupe cette année quantifie la structure des protéines dans une séquence de jetons structurels et l'introduit dans le modèle Transformer avec la séquence d'acides aminés, fusionnant ces deux types d'informations via un mécanisme d'attention distinct. Un autre exemple est le modèle contemporain ESM-3, qui prend en compte des informations plus riches, notamment le type d'acide aminé, la structure tertiaire complète, le jeton de structure tertiaire, la structure secondaire, la surface accessible au solvant (SASA) et la description fonctionnelle des protéines et des résidus, soit un total de 7 entrées.
Prédiction de la condition physique non supervisée et supervisée
Ensuite, nous discuterons du problème de prédiction de la condition physique.Étant donné que PLM peut modéliser la distribution de probabilité des séquences de protéines, il peut être directement utilisé pour la prédiction de la fitness des mutations sans données étiquetées. Cette méthode est appelée prédiction zéro coup ou prédiction non supervisée.

Plus précisément, le PLM évalue les mutations en calculant le rapport de vraisemblance logarithmique entre le mutant et le type sauvage. Pour le modèle autorégressif, la probabilité de la séquence P est le produit des probabilités de générer chaque acide aminé. Le score de mutation peut être obtenu en soustrayant le logP de type sauvage du logP mutant. Intuitivement parlant, il s’agit de comparer la probabilité d’apparition d’une mutation par rapport au type sauvage, puis d’évaluer l’impact de la mutation. Il s’agit d’une méthode d’évaluation empirique.
Pour le modèle de masquage, il est impossible de calculer directement la probabilité de la séquence entière, mais il peut d'abord masquer un certain point, puis estimer la distribution de probabilité des acides aminés à ce point. Par conséquent, pour chaque position de mutation, le logP de l'acide aminé de type sauvage peut être soustrait du logP de l'acide aminé mutant prédit après masquage, puis la différence à toutes les positions peut être ajoutée pour obtenir le score du mutant.
De plus, comme les PLM fournissent des représentations vectorielles de séquences protéiques, ils peuvent également être affinés pour obtenir une prédiction de fitness supervisée lorsqu'il existe suffisamment de données expérimentales.
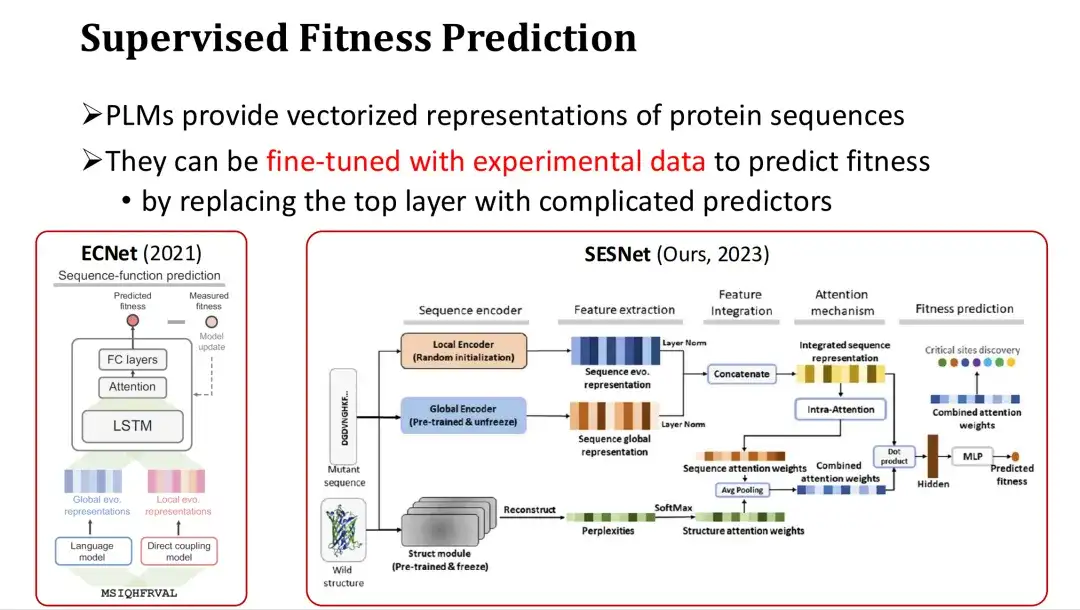
L'approche spécifique consiste à ajouter une couche de sortie pour prédire la forme physique (comme un mécanisme d'attention ou un perceptron multicouche MLP) après la dernière couche de fonctionnalités du PLM, et à utiliser l'étiquette de forme physique pour une formation complète ou partielle. Par exemple, ECNet ajoute des fonctionnalités MSA basées sur les fonctionnalités du grand modèle, les intègre via LSTM et effectue une formation supervisée. Le modèle SESNet développé par notre groupe de recherche l'année dernière combine les caractéristiques de séquence d'ESM-1b, les caractéristiques structurelles d'ESM-IF et les caractéristiques MSA pour effectuer une prédiction de fitness supervisée.
Introduction à FSFP : un petit exemple de méthode d'apprentissage pour PLM
Importance de l'apprentissage par petits échantillons pour la prédiction de la condition physique
Avant d’introduire la méthode FSFP, il est nécessaire de clarifier l’importance de l’apprentissage sur de petits échantillons dans la prédiction de la condition physique. Bien que les méthodes non supervisées ne nécessitent pas de données étiquetées pour la formation, leur précision de notation à zéro coup est plus faible. De plus, comme les scores basés sur les rapports de vraisemblance logarithmique ne peuvent refléter que certaines lois naturelles des protéines, il est également difficile de prédire efficacement les propriétés non naturelles des protéines.

D’autre part, bien que les méthodes d’apprentissage supervisé soient précises, en raison du grand nombre de paramètres PLM, elles nécessitent des données expérimentales à grande échelle pour la formation afin d’améliorer considérablement les performances. L'évaluation des modèles d'apprentissage supervisé implique généralement de diviser l'ensemble de données à haut débit existant en 8:2, tandis que l'ensemble d'entraînement de 80% peut déjà contenir des dizaines de milliers de données, ce qui est très coûteux à obtenir en pratique.
Pour résoudre ce problème, nous proposons la méthode FSFP, une approche d’apprentissage par petites étapes pour le PLM. Cette méthode peut améliorer considérablement les performances de prédiction de fitness du PLM en utilisant un petit nombre d'échantillons d'entraînement (des dizaines). Dans le même temps, la méthode FSFP est très flexible et peut être appliquée à différents PLM.
Méthode FSFP : apprentissage par classement pour la condition physique
Les méthodes d’apprentissage supervisé précédentes ont toutes considéré la prédiction de la condition physique comme un problème de régression, c’est-à-dire l’optimisation du modèle en calculant l’erreur quadratique moyenne (MSE) entre la sortie du modèle et l’étiquette de condition physique. Cependant, dans des conditions d’échantillon réduit, le modèle de régression est très facile à surajuster et la perte d’apprentissage diminue très rapidement. Nous avons donc changé notre façon de penser et n’avons pas fait de régression, mais plutôt un apprentissage par classement, qui ne nécessitait qu’un tri précis et ne nécessitait pas d’ajustement précis des valeurs numériques.
Cette approche présente deux avantages majeurs. Tout d’abord, le séquençage lui-même répond aux besoins fondamentaux de l’ingénierie des protéines, qui nécessite uniquement de mesurer l’efficacité relative des mutations. Deuxièmement, la tâche de classement est plus simple que la prédiction de valeurs absolues.
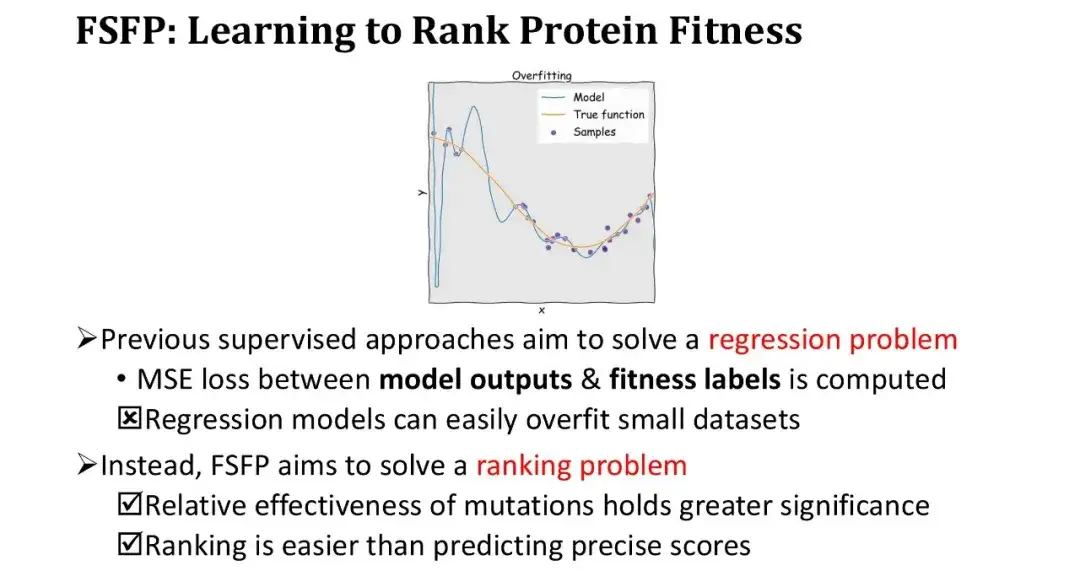
Dans l'itération de formation, nous trions l'ensemble échantillonné de mutants dans l'ordre inverse en fonction de leurs étiquettes, puis calculons la perte de classement - ListMLE en fonction des valeurs de prédiction du modèle pour ces mutants.Plus le classement des valeurs prédites du modèle est proche du classement réel, plus la perte est faible. Parmi eux, nous utilisons la fonction de notation à zéro coup basée sur le rapport de vraisemblance logarithmique comme fonction de notation f pour le modèle à mutation. L'objectif est d'utiliser le score zéro-shot comme point de départ et de le corriger progressivement avec les données d'entraînement pour améliorer les performances sans réinitialiser un module, réduisant ainsi la difficulté de l'entraînement.
Méthode FSFP : réglage fin efficace des paramètres du PLM
Étant donné que le nombre de paramètres dans un PLM est généralement aussi élevé que des centaines de millions, le réglage précis de l'ensemble du modèle avec très peu de données conduira inévitablement à un surajustement.Nous avons donc introduit une deuxième technique, LoRA, pour limiter le nombre de paramètres entraînables du modèle.
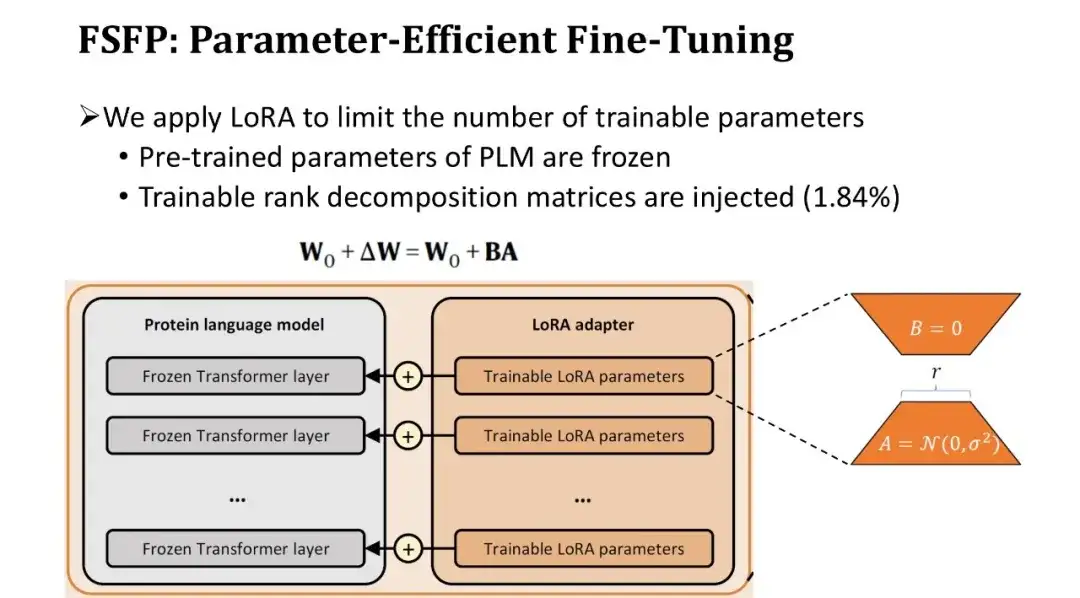
LoRA insère une paire de matrices de décomposition de rang entraînables dans la couche entièrement connectée de chaque bloc de Transformer, en gardant les paramètres pré-entraînés inchangés. Étant donné que la matrice de décomposition des rangs est très petite, le nombre de paramètres pouvant être formés peut être réduit à 1,84% d'origine. Bien que le nombre de paramètres pouvant être formés soit réduit, la capacité d'apprentissage du modèle est toujours garantie car chaque couche du Transformer est affinée.
Méthode FSFP : application du méta-apprentissage à la prédiction de la condition physique
Pour éviter le surapprentissage, nous avons non seulement utilisé une meilleure fonction de perte, mais nous avons également limité la quantité de paramètres pouvant être formés grâce à la technologie LoRA. Cependant, même dans ce cas, il existe toujours un risque de surapprentissage si trop d’itérations d’entraînement sont effectuées sur de petits échantillons de données d’entraînement. Nous espérons donc améliorer rapidement les performances du modèle avec moins d’itérations de formation.Sur la base de ce besoin, nous avons adopté la troisième technologie : le méta-apprentissage. L'idée de base du méta-apprentissage est de laisser d'abord le modèle accumuler de l'expérience sur certaines tâches auxiliaires pour obtenir un modèle initial, puis d'utiliser le modèle initial pour s'adapter rapidement à de nouvelles tâches.
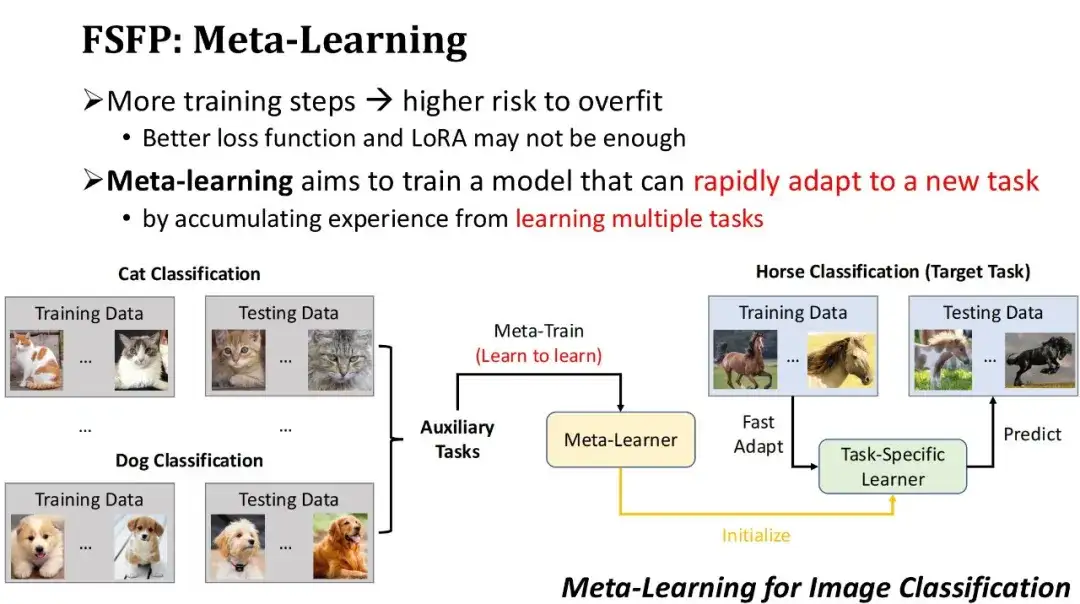
Comme le montre la figure ci-dessous, il s’agit d’un exemple de classification d’images basée sur le méta-apprentissage. Supposons que la tâche cible soit de former un modèle pour classer les chevaux, mais qu'il existe relativement peu de données étiquetées sur les chevaux. Par conséquent, nous pouvons d'abord trouver certaines tâches auxiliaires avec une grande quantité de données, telles que la classification des chats, la classification des chiens, etc., et utiliser des algorithmes de méta-apprentissage pour nous entraîner sur ces tâches auxiliaires, apprendre à apprendre de nouvelles tâches et obtenir un méta-apprenant. Ensuite, en utilisant ce méta-apprenant comme modèle initial et en l'entraînant pendant plusieurs étapes avec une petite quantité de données de chevaux étiquetées, nous pouvons rapidement obtenir un classificateur de chevaux. De toute évidence, la condition préalable pour que le méta-apprentissage fonctionne est que les tâches auxiliaires utilisées soient suffisamment proches des tâches cibles.
Comment appliquer le méta-apprentissage au scénario de prédiction de la condition physique ?Tout d’abord, notre objectif est de classer les mutations de la protéine cible par Fitness, et le modèle à entraîner est PLM utilisant la technologie LoRA.
Nous adoptons deux stratégies pour construire des tâches auxiliaires. La première étape consiste à rechercher des ensembles de données d’expériences de mutation de protéines similaires dans la base de données DMS existante en fonction de la similitude avec la protéine cible, et à sélectionner les deux premiers ensembles de données comme deux tâches auxiliaires.Le point de départ pour y parvenir est de considérer que le paysage de fitness de protéines similaires est également similaire.
La deuxième stratégie consiste à utiliser le modèle MSA pour évaluer les mutations candidates de la protéine cible afin de former un ensemble de données de pseudo-étiquettes et de l'utiliser comme troisième tâche auxiliaire.La raison pour laquelle nous avons choisi le modèle MSA est que l’effet de prédiction de mutation du modèle MSA n’est généralement pas inférieur à celui du PLM. Nous espérons pouvoir améliorer les données grâce à MSA et exploiter pleinement la capacité de représentation du PLM.
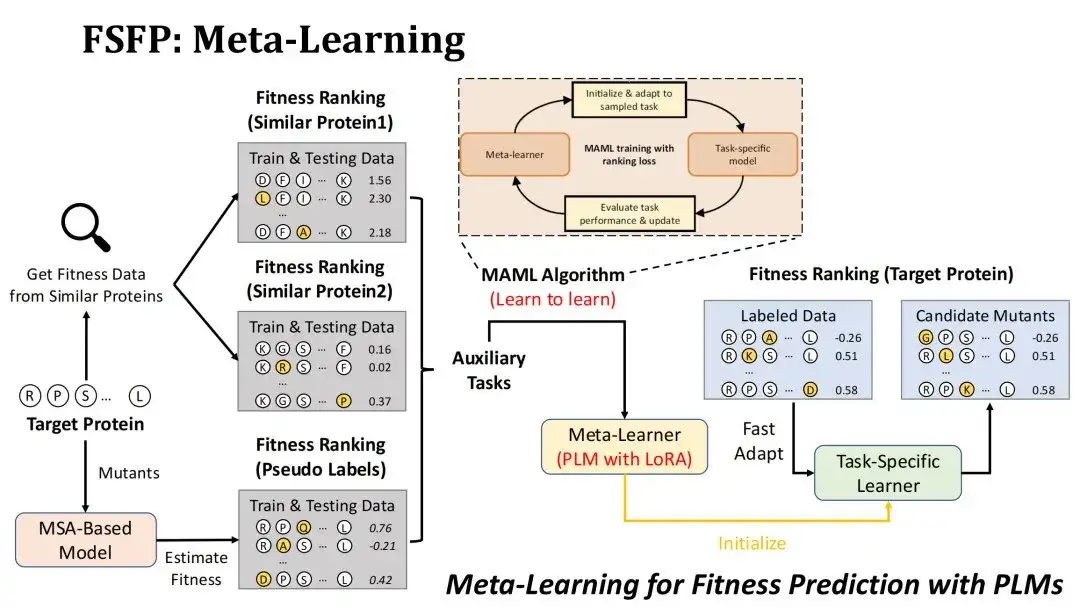
L'algorithme de méta-apprentissage que nous utilisons est MAML, dont l'objectif d'entraînement est de rendre la perte de test du méta-apprenant aussi petite que possible après avoir peaufiné k étapes avec les données d'entraînement d'une tâche auxiliaire, afin qu'elle puisse converger approximativement après avoir peaufiné k étapes sur la tâche cible.
Évaluation des performances de la méthode FSFP dans la prédiction de la fitness des protéines
Création de benchmarks
Nos données de référence proviennent de ProteinGym, qui contenait à l'origine 87 ensembles de données DMS et a maintenant été mis à jour à 217.Les protéines correspondant à 87 DMS sont grossièrement divisées en quatre catégories : eucaryotes, procaryotes, humains et virus, couvrant un total d'environ 15 millions de mutations et de fitness correspondant.
Pour chaque ensemble de données, nous avons sélectionné au hasard 20, 40, 60, 80 et 100 mutations ponctuelles comme petits ensembles d'entraînement d'échantillons, et les mutations restantes ont été utilisées comme ensembles de test. Il convient de noter que nous n’avons pas utilisé d’ensemble de validation supplémentaire pour l’arrêt précoce, mais avons plutôt estimé le nombre d’étapes de formation par validation croisée sur l’ensemble de formation.

Il a été mentionné précédemment que le méta-apprentissage nécessite trois tâches auxiliaires, dont deux sont récupérées dans la base de données DMS en fonction de leur similarité avec la protéine cible.Lors de l'entraînement sur un ensemble de données, nous récupérons les données du reste des ensembles de données dans ProteinGym, en supposant qu'il s'agit de la base de données.
Comme le montre la figure ci-dessous à droite, chaque protéine de ProteinGym est utilisée comme requête, et la distribution de similarité des protéines les plus similaires est récupérée respectivement via MMseqs2 et FoldSeek. On peut constater que la séquence moyenne ou la similarité structurelle des protéines les plus similaires est d’environ 0,5. La troisième tâche auxiliaire consiste à évaluer les mutations à l’aide du modèle MSA. Nous avons choisi le modèle GEMME, qui construit un arbre évolutif basé sur MSA et calcule la conservation de chaque point de l'arbre évolutif pour noter la mutation.
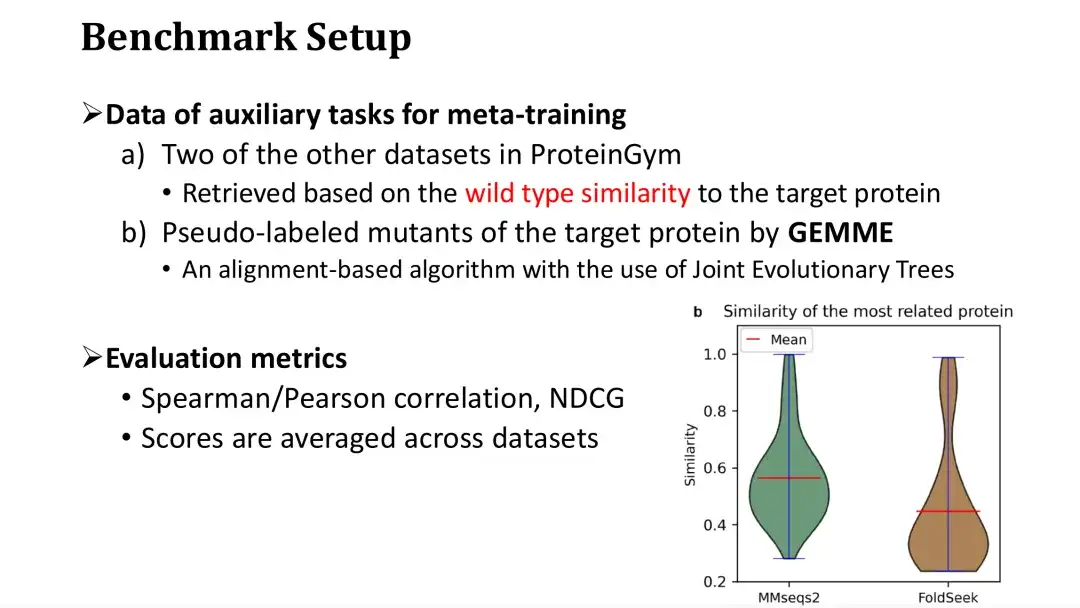
Les indicateurs d’évaluation utilisés sont le coefficient de Spearman/Pearson et le NDCG, qui sont des critères d’évaluation courants dans les tâches de prédiction de la condition physique. Le score d’évaluation final est le score moyen sur les 87 ensembles de données.
Expérience d'ablation de FSFP sur ESM-2
Comme le montre la figure ci-dessous, l’axe des x dans la figure de gauche représente la taille de l’ensemble d’entraînement, l’axe des y représente le coefficient de Spearman et chaque ligne correspond à une configuration de modèle différente. La ligne supérieure représente le modèle FSFP complet ; la deuxième ligne représente le remplacement de la troisième tâche auxiliaire de méta-apprentissage par des données DMS de protéines similaires sans utiliser MSA. On peut voir que les performances du modèle diminuent après la suppression des informations MSA ; la troisième ligne représente la non-utilisation du méta-apprentissage, s'appuyant uniquement sur l'apprentissage par classement et LoRA, et le coefficient de Spearman diminue encore.
La ligne verte représente le modèle de régression de crête précédemment publié dans NBT, qui est l’un des rares modèles de base actuellement adaptés aux scénarios de petits échantillons ; la ligne pointillée grise représente le score zéro coup de l'ESM-2 ; les deux lignes inférieures représentent les résultats de la formation ESM-2 à l'aide de méthodes de régression traditionnelles.
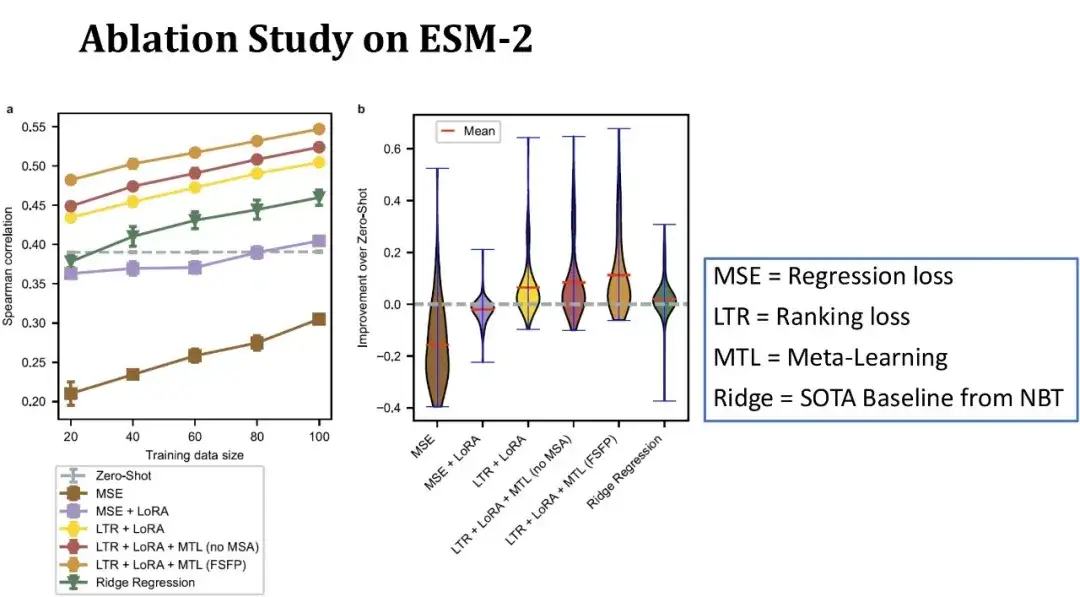
Globalement, lorsqu’il n’y a que 20 échantillons d’entraînement, notre méthode améliore le Spearman de 10 points par rapport au zéro-shot, et chaque module joue un rôle positif dans les performances du modèle. La figure de droite montre la distribution de l’amélioration des performances par rapport au zero-shot sur 87 ensembles de données, avec une taille d’ensemble d’entraînement de 40 échantillons.On peut constater que notre méthode peut améliorer les performances du modèle sur la plupart des ensembles de données, et l’amélioration sur certains ensembles de données dépasse même 40 points, montrant des performances plus stables que la ligne de base.
Efficacité du méta-apprentissage
L’objectif du méta-apprentissage est de permettre au PLM de converger rapidement vers la tâche cible avec un petit nombre d’itérations.Voici quelques exemples pour illustrer cela.
Les 3 graphiques suivants montrent les courbes d’entraînement du réglage fin sur 3 ensembles de données utilisant 40 échantillons d’entraînement. L'axe des x représente le nombre d'étapes d'entraînement et l'axe des y représente le coefficient de Spearman sur l'ensemble de test. Les lignes orange et rouge en haut sont toutes deux des modèles entraînés avec le méta-apprentissage, la première utilise MSA pour construire des tâches auxiliaires, tandis que la seconde ne le fait pas. La ligne jaune représente le modèle utilisant uniquement l'apprentissage du classement et LoRA sans méta-apprentissage.
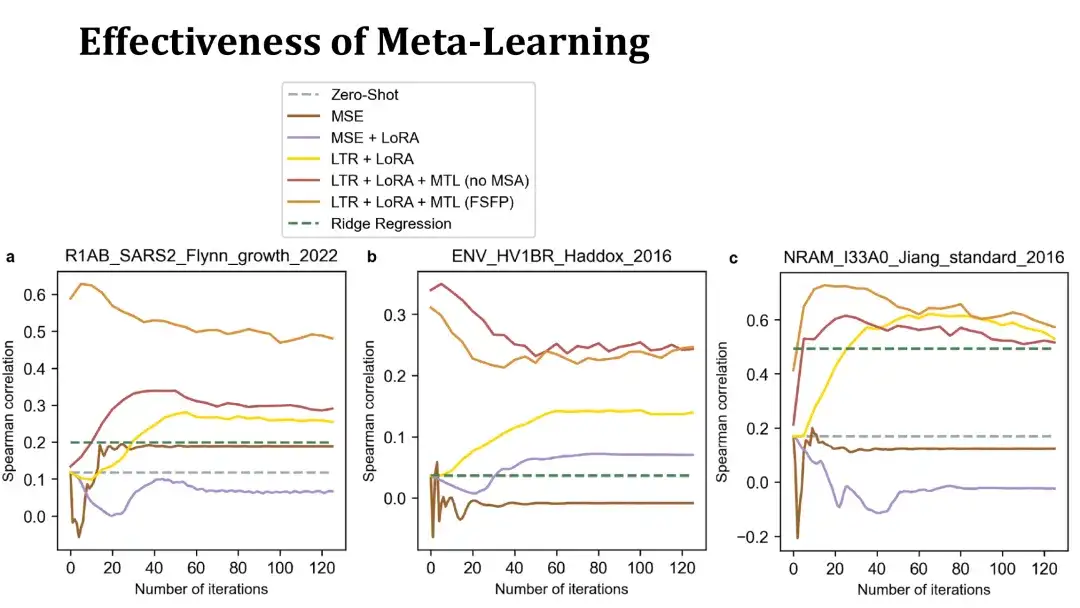
Comme vous pouvez le voir,Les modèles formés avec le méta-apprentissage peuvent améliorer les performances sur les protéines cibles plus rapidement et atteindre des scores plus élevés en 20 étapes, surpassant parfois même le modèle initial sans réglage fin. Cela indique que le méta-apprentissage produit un modèle initial efficace.Le modèle basé sur MSE ci-dessous fonctionne mal et s'adapte rapidement, ce qui rend difficile de dépasser la méthode du zéro coup.
Résultats de l'application du FSFP à différents PLM
Nous avons sélectionné trois PLM typiques, à savoir ESM-1v, ESM-2 et SaProt.Les deux premiers modèles utilisent uniquement les informations de séquence protéique, tandis que SaProt combine les jetons de structure tertiaire des protéines.
Le graphique linéaire de gauche montre le score de Spearman pour prédire l’effet d’une mutation ponctuelle unique sous différentes tailles d’ensemble d’entraînement. La même couleur représente le même modèle et différentes formes de points représentent différentes méthodes d’entraînement. Les points ci-dessus représentent la méthode FSFP, le triangle inversé ci-dessous représente la régression de crête et la ligne pointillée représente les performances zéro-shot du modèle. La ligne violette représente le modèle GEMME, qui n'est pas un PLM, mais la méthode de régression de crête peut être combinée avec lui.On peut voir que la méthode FSFP peut améliorer régulièrement les performances de chaque PLM, et est bien meilleure que la régression de crête et le tir zéro du modèle correspondant.
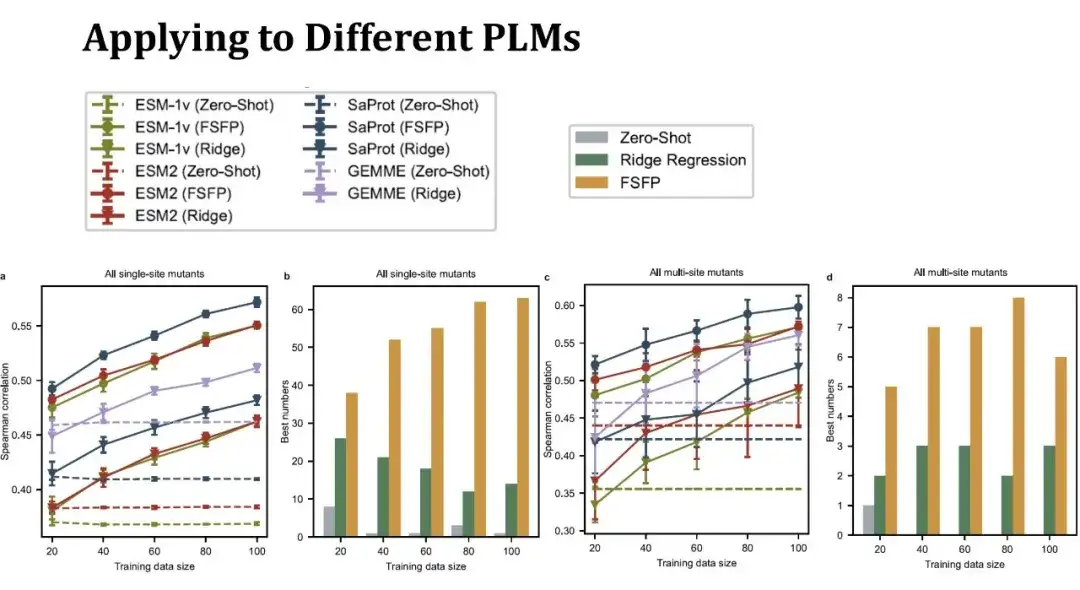
Le deuxième graphique à barres montre le nombre de scores les plus élevés obtenus en utilisant les trois stratégies (zéro-tir, régression de crête et FSFP) sur différents ensembles de données. Le FSFP fonctionne mieux dans la plupart des ensembles de données.Les deux figures de droite montrent les performances de prédiction des mutations multipoints. Il existe 11 ensembles de données de mutations multipoints impliqués, et les conclusions obtenues sont similaires à celles des mutations à point unique. Cependant, le modèle de régression de crête présente ici une variance plus importante, ce qui indique qu’il est sensible à la division des données.
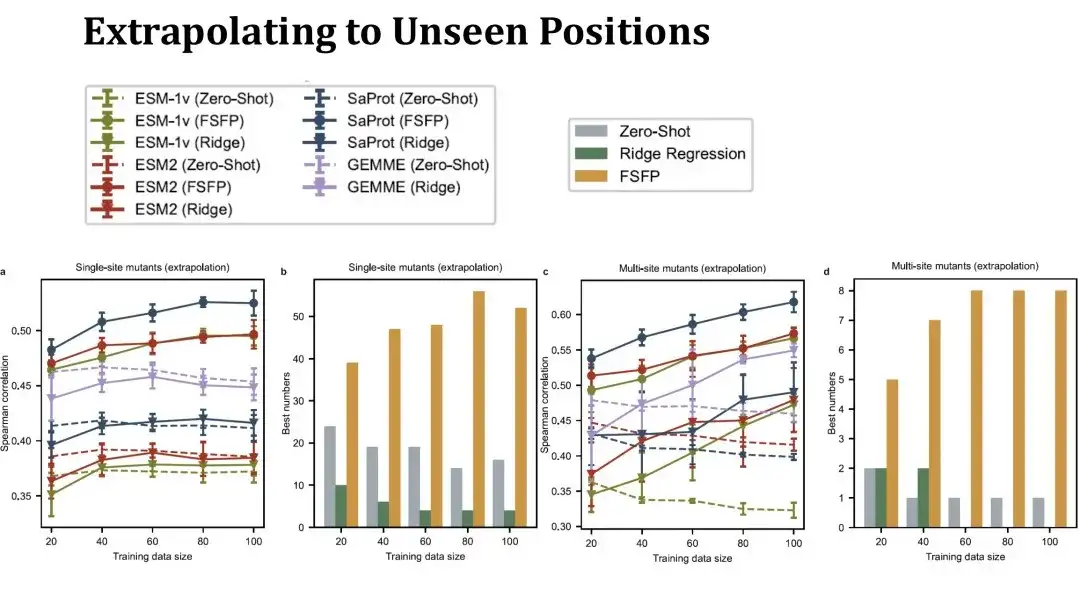
Nous avons ensuite évalué les performances d’extrapolation du FSFP, en évaluant spécifiquement les performances de prédiction sur des sites de mutation non observés dans l’ensemble d’entraînement.. Dans ce cas, l'ensemble de test sera beaucoup plus petit qu'auparavant et l'ensemble de test changera considérablement à mesure que l'ensemble d'entraînement deviendra plus grand, de sorte que les performances zéro-shot dans le tableau ne sont plus une ligne droite. Ce paramètre est plus difficile. Nous pouvons voir que les performances de la régression de crête de mutation à point unique sur la gauche peuvent difficilement dépasser le tir zéro, mais le FSFP peut toujours améliorer régulièrement les performances. Les résultats des tests de mutations multipoints à droite montrent également que notre méthode de formation a une bonne capacité de généralisation.
Transformer Phi29 avec FSFP
De plus, nous avons également utilisé le FSFP pour réaliser une étude de cas sur la modification des protéines.La protéine cible est Phi29, une ADN polymérase, et nous espérons améliorer sa Tm par mutation ponctuelle.
Le processus expérimental est le suivant : tout d'abord, utiliser ESM-1v pour effectuer un score zéro sur les mutations ponctuelles saturées, sélectionner les 20 principales mutations et effectuer des expériences humides pour mesurer Tm ; utilisez ensuite ces 20 données expérimentales comme ensemble d'entraînement, utilisez FSFP pour entraîner ESM-1v, utilisez le modèle entraîné pour noter à nouveau les mutations ponctuelles saturées et resélectionnez les 20 principales mutations pour les tests.
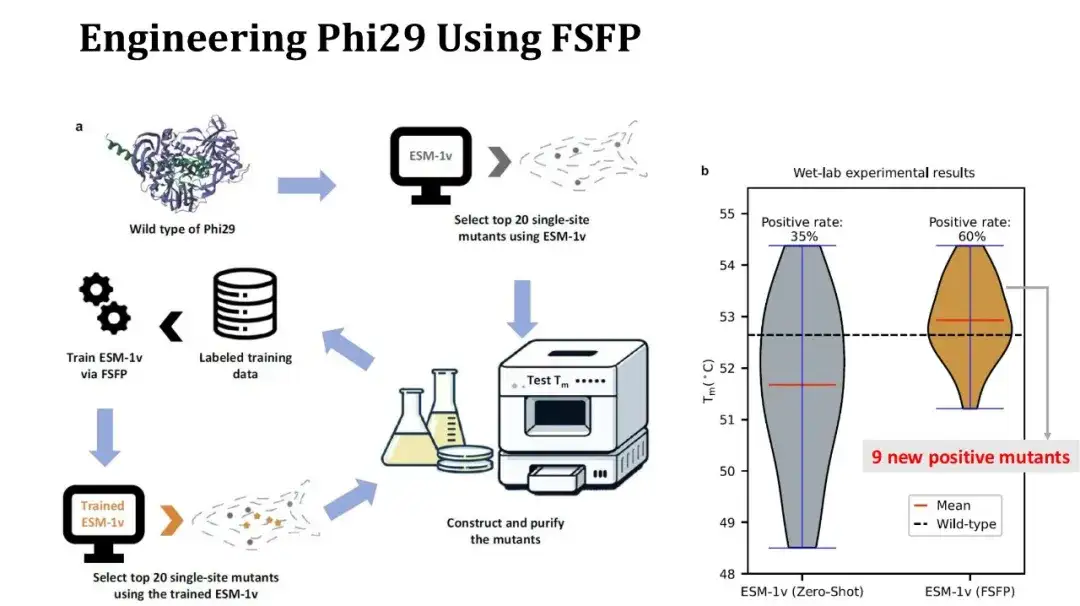
La figure de droite montre la comparaison des distributions de Tm avant et après les deux séries d’expériences. Sept mutations sur 20 au premier tour étaient positives, ce chiffre est passé à 12 au deuxième tour, et le Tm moyen a augmenté de 1 degré. Parmi elles, 9 des mutations positives trouvées au deuxième tour étaient nouvelles. Bien que le taux positif et le Tm moyen aient été améliorés, malheureusement le Tm le plus élevé n'a pas augmenté car la mutation avec le Tm le plus élevé obtenu au deuxième tour existait toujours dans les résultats du premier tour. Cependant, étant donné que davantage de mutations ponctuelles positives ont été obtenues, nous pouvons essayer de combiner ces sites pour mener des expériences de mutation ponctuelle élevée afin d'améliorer encore Tm.
Résumé de la méthode FSFP et perspectives de recherche futures
FSFP est une stratégie d'apprentissage de petits échantillons pour PLM, qui peut améliorer considérablement les performances du PLM dans la prédiction de l'effet de mutation en utilisant un petit nombre (des dizaines) d'échantillons d'entraînement étiquetés, et peut être appliquée de manière flexible à une variété de PLM différents.Les expériences montrent que la conception du FSFP est raisonnable :
* L’apprentissage par classement répond à l’exigence de base du classement des mutations dans l’ingénierie des protéines et réduit la difficulté de la formation ;
* LoRA réduit le risque de surapprentissage en contrôlant la quantité de paramètres entraînables du PLM ;
* Le méta-apprentissage peut fournir de bons paramètres initiaux pour le modèle, permettant au modèle de migrer rapidement vers la tâche cible.

Enfin, nous discutons des orientations futures de l’évolution dirigée assistée par l’IA. Le processus général de l'évolution dirigée assistée par l'IA consiste à commencer par un ensemble de mutations initiales, à obtenir leurs étiquettes de fitness grâce à des expériences humides et à utiliser les données étiquetées renvoyées par les expériences pour former le modèle d'apprentissage automatique. Ensuite, la prochaine série de mutations à tester est sélectionnée en fonction des prédictions du modèle, et le processus est répété.
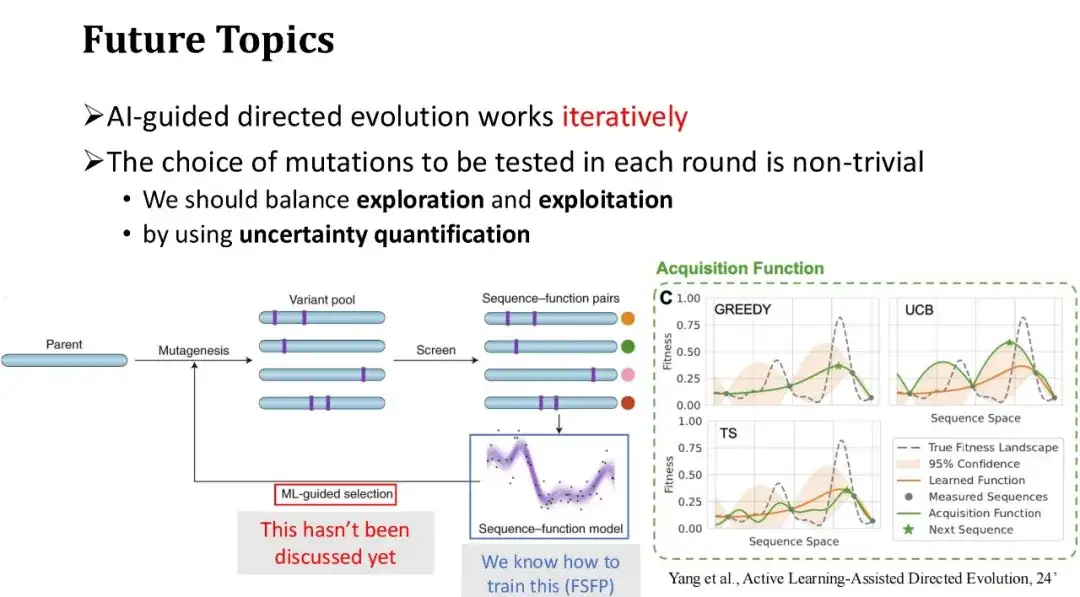
FSFP résout principalement le problème de la formation de petits échantillons du modèle à chaque cycle d'itération expérimentale et améliore la précision de prédiction du modèle.Cependant, nous n’avons pas encore discuté de la manière de sélectionner efficacement les mutations à tester lors du prochain tour, c’est-à-dire les nouveaux échantillons d’entraînement à ajouter lors du prochain tour. Dans l’exemple précédent de modification de la protéine Phi29, nous avons directement sélectionné les 20 principales mutations avec les scores de modèle les plus élevés. Cependant, dans un scénario itératif à plusieurs tours, la stratégie de sélection gourmande n’est pas nécessairement la meilleure méthode, car elle a tendance à tomber dans l’optimalité locale. Il faut donc trouver un équilibre entre l’exploration et l’exploitation.
En fait, le processus de sélection itérative d’échantillons de test à étiqueter et d’expansion progressive des données de formation est un problème d’apprentissage actif, qui a fait progresser la recherche dans le domaine de l’ingénierie des protéines. Par exemple, Frances H. Arnold, une autorité en matière d’évolution dirigée, aborde des questions connexes dans son article « Active Learning-Assisted Directed Evolution ».
Adresse du document :
https://www.biorxiv.org/content/10.1101/2024.07.27.605457v1.full.pdf
Nous pouvons utiliser des techniques de quantification de l’incertitude pour évaluer l’incertitude du score du modèle pour chaque mutant. Sur la base de ces incertitudes, la stratégie de sélection des échantillons d’essai sera plus diversifiée.. Une stratégie couramment utilisée est la méthode UCB, qui sélectionne les échantillons de mutation avec l’incertitude de prédiction du modèle la plus élevée pour le prochain cycle d’annotation, c’est-à-dire qu’elle donne la priorité aux échantillons avec la plus grande variance de prédiction. C’est similaire au processus d’apprentissage humain : si nous ne maîtrisons pas bien certains points de connaissance ou si nous sommes incertains à leur sujet, nous nous concentrerons sur le renforcement de notre apprentissage.