Command Palette
Search for a command to run...
Nouveaux Résultats De La Revue De Référence Cell Discovery ! L'équipe Dirigée Par Hong Liang De l'Université Jiao Tong De Shanghai a Proposé Un Modèle CPDiffusion Pour La Conception ultra-faible Coût Et Entièrement Automatique De Protéines Fonctionnelles
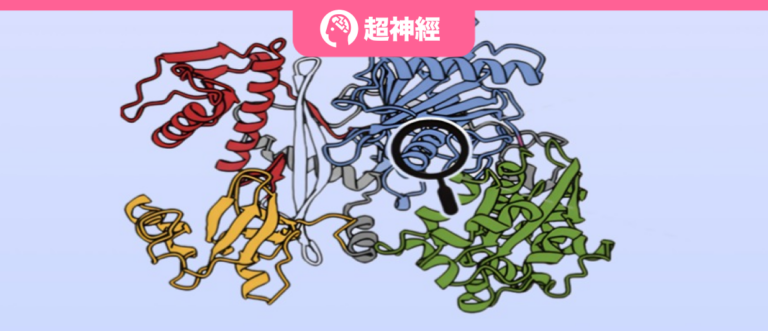
Les protéines sont les principaux exécutants des activités de la vie, et la relation entre leur structure et leur fonction a toujours été un sujet de recherche essentiel dans le domaine des sciences de la vie. Ces dernières années, avec l'essor de l'apprentissage en profondeur, avec ses puissantes capacités de traitement de données, le modèle peut apprendre la relation de cartographie entre la séquence protéique, la structure et sa fonction, et concevoir de nouvelles protéines avec une stabilité plus élevée, une affinité de liaison plus forte et une activité enzymatique plus élevée, ce qui peut considérablement améliorer l'efficacité de la conception des protéines et réduire efficacement ses coûts de R&D.
Cependant, les méthodes existantes nécessitent généralement la formation d’un modèle avec des paramètres extrêmement importants sur un ensemble de données à grande échelle, ce qui est difficile à généraliser à des protéines spécifiques avec des séquences homologues rares, et ne peut souvent générer que des protéines avec des structures et des fonctions relativement simples. De plus, la vérification expérimentale montre que les protéines conçues ont généralement une activité plus faible et que celles qui peuvent surpasser les protéines de type sauvage sont rares.
À cet égard, Zhou Bingxin, chercheur adjoint au sein du groupe de recherche de Hong Liang de l'École des sciences naturelles/École de physique et d'astronomie/Institut d'études avancées de Zhangjiang/École de pharmacie de l'Université Jiao Tong de Shanghai, et d'autres ont conçu un cadre de modèle de probabilité de diffusion CPDiffusion.Ce cadre combine plusieurs conditions de génération telles que la structure du squelette protéique et les sites actifs, et peut apprendre la relation de mappage implicite entre la séquence protéique, la structure et la fonction à un coût de formation et de données très faible, puis générer diverses séquences protéiques. Ces séquences générées peuvent passer le test avec un taux de réussite très élevé lors de la vérification des expériences humides.
Il convient de noter que le processus de formation et d’inférence de CPDiffusion ne nécessite pratiquement aucune orientation d’expert.Il peut identifier automatiquement les régions hautement conservées, puis introduire davantage de changements dans les régions non conservées en fonction des fonctions des régions conservées pour augmenter la diversité des séquences générées. L'étude, intitulée « Un modèle de diffusion conditionnelle de protéines génère des séquences d'endonucléases programmables artificielles avec une activité améliorée », a été publiée dans Nature's Cell Discovery.
Points saillants de la recherche :
* L'étude a conçu et généré avec succès les endonucléases KmAgo et PfAgo, dont l'activité de clivage de l'ADN a été augmentée de plus de 10 fois, significativement plus élevée que l'activité des protéines de type sauvage à température moyenne actuellement découvertes
* Cette étude peut modifier des centaines d'acides aminés à la fois, offrant ainsi davantage de possibilités pour la recherche en ingénierie des protéines
* La génération diversifiée de nouvelles séquences protéiques peut également élargir la base de données des familles de protéines, fournissant aux scientifiques des ressources de recherche plus riches

Lien vers l'article :
https://www.nature.com/articles/s41421-024-00728-2
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : garantir la diversité des échantillons et éviter les biais de données
Afin d'apprendre la relation de cartographie entre la séquence protéique, la structure et la fonction,Le modèle CPDiffusion a été formé avec 20 000 protéines de type sauvage de CATH 4.2. De plus, les chercheurs ont ajouté 694 protéines pAgos à l'ensemble d'entraînement pour améliorer la compréhension du modèle des caractéristiques des protéines à générer.
Ces protéines proviennent de la famille de protéines pAgo compilée dans des études précédentes, comprenant des protéines pAgo courtes, longues-A et longues-B, garantissant la diversité des échantillons sélectionnés pour réduire les éventuels problèmes de biais de données. De plus, la plupart des protéines WT de l'ensemble de données sont des pAgos mésophiles, et seules quelques protéines pAgo à long A sont thermophiles.
Architecture du modèle : conception automatisée de protéines pAgo en 6 étapes
Pour vérifier l’effet de la CPDiffusion sur la génération de protéines fonctionnelles, les chercheurs ont choisi de se concentrer sur la protéine pAgo. La protéine pAgo est une endonucléase qui joue un rôle important dans le processus d'interférence de l'ADN des procaryotes. Il peut reconnaître et couper spécifiquement des séquences d'ADN ou d'ARN monocaténaires spécifiques et présente une large valeur d'application dans le domaine du diagnostic. De plus, les protéines pAgo ont une forte affinité pour les substrats et peuvent reconnaître spécifiquement les séquences cibles, ce qui en fait des outils importants pour l'imagerie et l'édition de gènes.
Les chercheurs ont utilisé le cadre CPDiffusion pour concevoir de nouvelles protéines pAgo.Comme le montre la figure a ci-dessous,Tout d’abord, la séquence et les informations de la protéine d’entrée (pAgo d’origine) sont converties en une représentation graphique qui affiche les propriétés biochimiques et topologiques moléculaires de la protéine au niveau des acides aminés.Comme le montre la figure b,La protéine entre dans la phase de diffusion directe, où chaque type d'acide aminé de la protéine d'origine suit une certaine matrice de probabilité de substitution et est détruit dans une série d'étapes (T étapes) jusqu'à ce que la séquence entière soit uniformément distribuée.
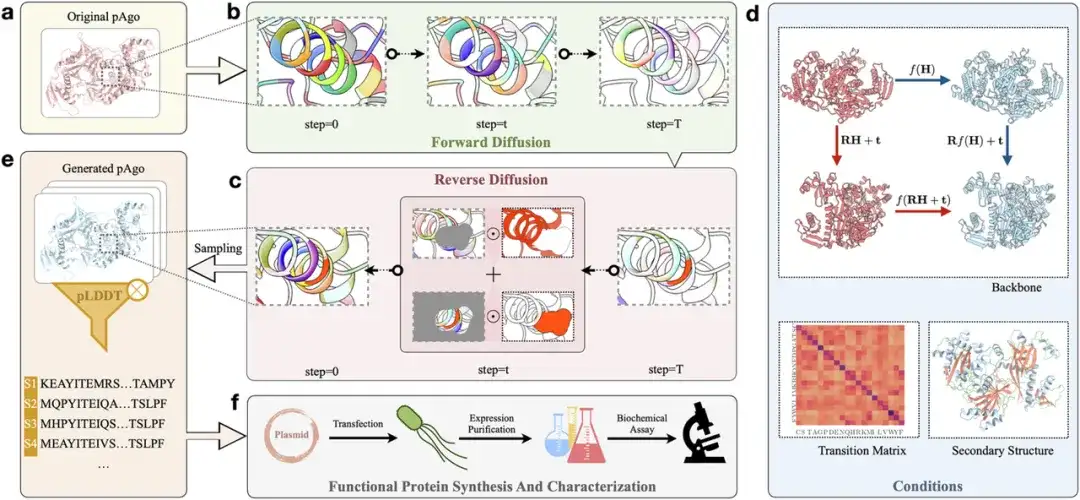
Comme le montre la figure c ci-dessus,Au cours de l'étape de diffusion inverse, les chercheurs ont échantillonné au hasard des acides aminés provenant de 20 types d'acides aminés répartis uniformément, puis ont progressivement débruité la séquence protéique.Comme le montre la figure d ci-dessus,Au cours du processus de débruitage, les chercheurs utilisent certaines conditions (telles que la structure du squelette de type sauvage de la protéine cible, la structure secondaire et la matrice de substitution d'acides aminés basée sur la protéine de type sauvage) pour guider le processus. Pour garantir que le modèle puisse apprendre l'équivariance implicite dans la structure tridimensionnelle de la protéine, les chercheurs ont utilisé une couche convolutionnelle de graphe équivariant pour ajuster la fonction de propagation. Le modèle génère ensuite une distribution de probabilité conjointe pour chaque position d’acide aminé sur le squelette protéique. En échantillonnant la distribution apprise, les chercheurs peuvent obtenir la séquence protéique correspondante (pAgo généré).Comme le montre la figure e ci-dessus.
Ensuite, les chercheurs ont utilisé AlphaFold2 pour effectuer une prédiction structurelle sur les séquences générées et ont sélectionné les séquences appropriées en évaluant des indicateurs tels que RMSD et pLDDT. enfin,Comme le montre la figure f ci-dessous,Ces séquences appropriées seront soumises à des expériences humides en laboratoire (synthèse, caractérisation et évaluation) pour confirmer davantage leurs propriétés réelles, telles que le niveau d'expression, l'activité enzymatique et la stabilité thermique.
Conclusion expérimentale : la nouvelle protéine présente une activité et une stabilité thermique plus fortes
Les chercheurs ont utilisé des protéines pAgo mésophiles (telles que KmAgo) et des protéines pAgo thermophiles (telles que PfAgo) comme protéines candidates et ont en outre généré deux groupes de nouvelles séquences protéiques. Comme le montre la figure ci-dessous, en utilisant le cadre de génération et de criblage CPDiffusion, les chercheurs ont généré avec succès 27 nouveaux KmAgos artificiels (Km-AP) et 15 nouveaux PfAgos artificiels (Pf-AP). Ces protéines nouvellement générées ont une identité de séquence de 50%-70% par rapport au modèle de type sauvage (WT) d'origine et une identité de séquence inférieure à 40% par rapport aux autres protéines WT non modèles (c'est-à-dire aux autres protéines WT dans la base de données NCBI).
* KmAgo est une enzyme mésophile avec une activité de clivage de l'ADN relativement faible dans le type sauvage, ce qui limite son potentiel dans les applications pratiques
* PfAgo est une enzyme ultra-thermogène. Le type sauvage a une activité de clivage de l'ADN plus élevée, mais il ne fonctionne généralement qu'à des températures élevées. À mesure que la température baisse, l’activité diminue également.
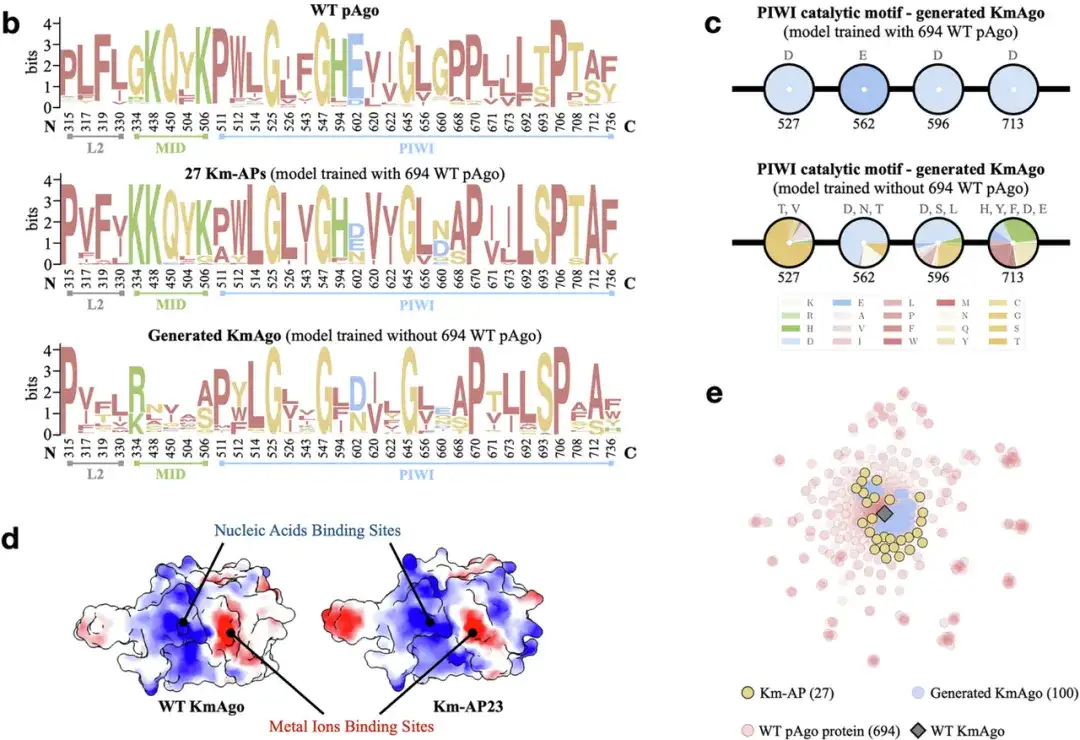
Il convient de mentionner queLe processus de formation et d’inférence de CPDiffusion ne nécessite pratiquement aucune orientation d’expert.Il peut identifier automatiquement les régions hautement conservées, introduisant ainsi davantage de changements dans les régions non conservées en fonction des fonctions des régions conservées pour augmenter la diversité des séquences générées.
Grâce à diverses vérifications expérimentales, comme le montre la figure ci-dessous, les chercheurs ont découvert que dans les nouvelles séquences générées pour KmAgo,Toutes les séquences ont été exprimées. Près de 901 nouvelles séquences TP3T présentaient une activité de clivage de l'ADN, et plus de 701 séquences TP3T présentaient une activité supérieure à celle du type sauvage. Parmi eux, le nouveau KmAgo le plus performant avait une activité près de 9 fois supérieure à celle du KmAgo de type sauvage. De plus, par rapport au KmAgo de type sauvage, la stabilité thermique de certains Km-AP a également été améliorée.
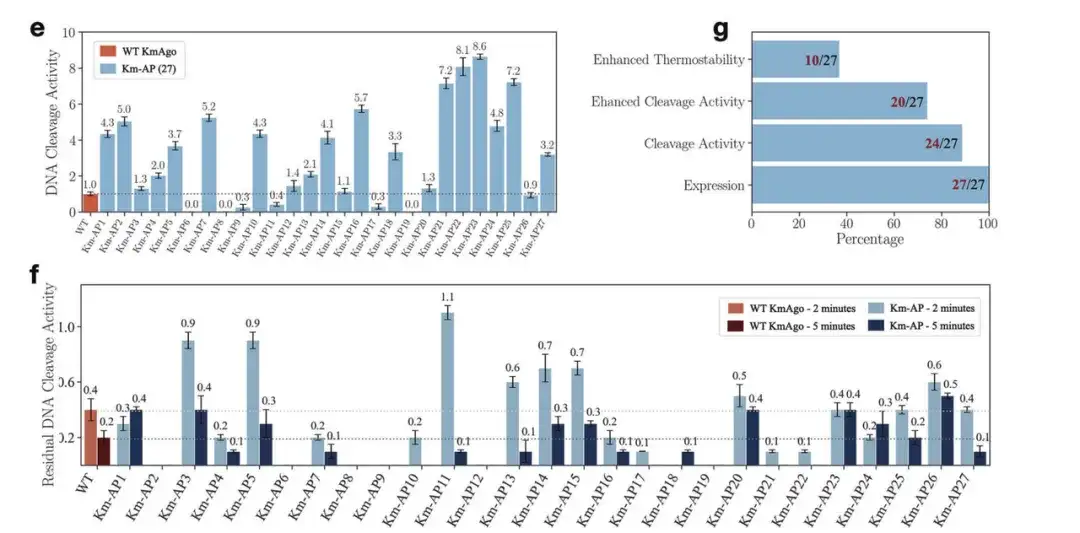
e : Activité de clivage de l'ADN de 27 Km-AP à 37 °C
g : Le nombre de protéines jouant différents rôles dans 27 Km-AP
f : Activités de clivage de l'ADN de WT KmAgo et 27 Km-AP après incubation à 42°C pendant 2 et 5 min.
Comme le montre la figure ci-dessous,Parmi les 15 nouvelles séquences générées pour PfAgo, toutes ont été exprimées et ont présenté une activité de clivage de l'ADN simple brin. Parmi eux, le nouveau PfAgo le plus performant a non seulement abaissé la température de fusion du PfAgo de type sauvage d'environ 100 °C à environ 50 °C, mais son activité de clivage de l'ADN simple brin à 45 °C était également deux fois supérieure à celle du PfAgo de type sauvage à 95 °C, et était 11 fois supérieure à celle du KmAgo de type sauvage à température moyenne.
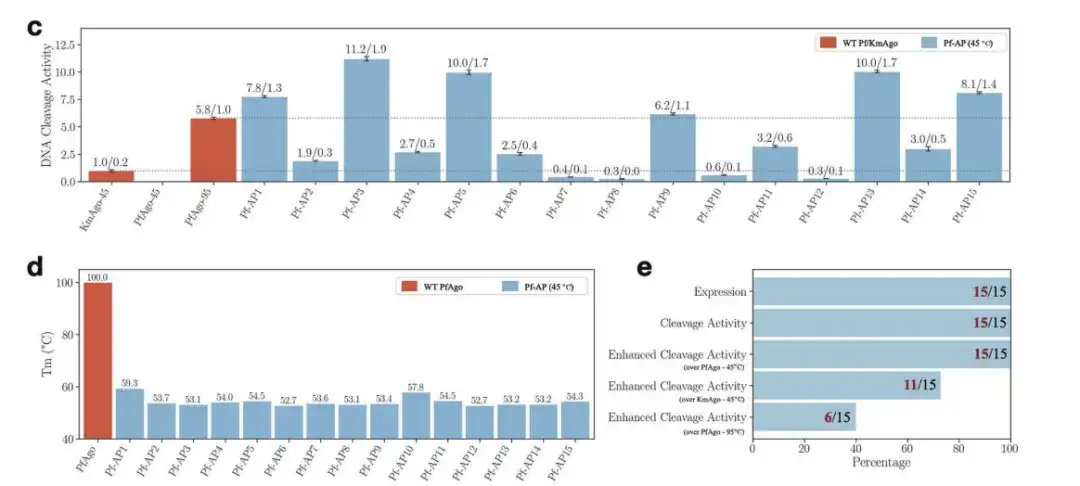
c : Activité de clivage de l'ADN de 15 Pf-AP à 45 °C
d : Température de fusion du PfAgo et du Pf-AP WT
e : Le nombre de séquences qui jouent des rôles différents parmi les 15 Pf-AP
En résumé, CPDiffusion peut être utilisé comme un nouvel outil puissant de conception de séquences protéiques capable d'apprendre automatiquement à partir de protéines fonctionnelles de type sauvage et de concevoir des séquences protéiques complexes plus puissantes, enrichissant la base de données protéiques existante et apportant davantage de possibilités à la conception de l'ingénierie des protéines.
L'IA remodèle l'avenir de l'ingénierie des protéines
L’utilisation de l’IA pour décoder les mystères des protéines est d’une importance capitale pour la numérisation de la recherche en sciences de la vie. Dans cette course à l’exploration de l’essence de la vie, les équipes de recherche chinoises progressent et contribuent continuellement. En tant que l’un des représentants les plus remarquables dans ce domaine,Le professeur Hong Liang, auteur correspondant de cette étude, et son équipe de recherche se concentrent depuis longtemps sur la modification dirigée par les protéines de l'IA et sur la conception assistée de médicaments.Les contenus de recherche spécifiques comprennent, sans s'y limiter, la prédiction et l'optimisation de la structure des protéines, la modification et la conception dirigées vers les protéines, la conception et l'optimisation de médicaments auxiliaires, etc. L'équipe a obtenu des résultats fructueux. Jusqu'à présent, un total de 77 articles ont été publiés, dont beaucoup ont été classés en tête de la revue Nature.
Page d'accueil du groupe de recherche du professeur Hong Liang :
https://ins.sjtu.edu.cn/people/lhong/index.html
Depuis 2021, l'équipe du professeur Hong Liang tente d'appliquer l'IA au domaine des protéines, par exemple,Créez des modèles propriétaires dans le domaine de l'ingénierie des protéines pour concevoir des séquences fonctionnelles de bout en bout.Ils ont collaboré avec le chercheur Tan Pan du Laboratoire d'intelligence artificielle de Shanghai pour proposer une méthode d'entraînement de réglage fin FSFP basée sur le modèle de pré-entraînement des protéines. Cette méthode peut entraîner efficacement le modèle de pré-entraînement des protéines en utilisant seulement 20 données expérimentales humides aléatoires, améliorant considérablement le taux de positivité de prédiction de mutation à point unique du modèle. Il peut être utilisé pour l’apprentissage sur de petits échantillons de l’adaptabilité des protéines et a montré un grand potentiel dans les applications pratiques.
L'équipe du professeur Hong Liang a également développé un réseau neuronal graphique sensible au microenvironnement appelé ProtLGN.Il peut apprendre et prédire les sites de mutation d'acides aminés bénéfiques à partir de la structure tridimensionnelle des protéines et guider la conception de mutations à site unique et de mutations multi-sites avec différentes fonctions. Les résultats expérimentaux ont montré que plus de 401 protéines mutantes à point unique conçues par TP3T ProtLGN ont surpassé leurs homologues de type sauvage.
Plus de détails : Sans données expérimentales pour guider l'évolution dirigée des protéines, le groupe de recherche de Hong Liang de l'Université Jiaotong de Shanghai a publié le réseau neuronal graphique sensible au microenvironnement ProtLGN
De plus, ils ont introduit un adaptateur SES-Adapter simple, efficace et évolutif,La combinaison d'intégrations de modèles de langage protéique avec des intégrations de séquences de structures pour créer des représentations tenant compte de la structure peut améliorer considérablement les performances des modèles de langage protéique.
La recherche ci-dessus démontre le puissant potentiel de l’apprentissage profond dans la conception des protéines. Il ne fait aucun doute qu’avec l’application plus poussée de la technologie d’apprentissage profond dans le domaine des protéines, la recherche en ingénierie des protéines ouvrira la voie à un espace de développement plus large.
Références :
https://mp.weixin.qq.com/s/a4gsV4yjzKnW4u6Vtl8LiQ
https://ins.sjtu.edu.cn/article








