Command Palette
Search for a command to run...
Sensibilité Améliorée Par 56%, CUHK/Fudan/Yale Et d'autres Ont Proposé Conjointement Une Nouvelle Méthode De Détection d'homologues Protéiques

Les protéines constituent la base matérielle de la vie et le principal vecteur des activités vitales. À l’ère post-génomique, avec le développement de la technologie de détermination des protéines, la taille des bases de données de séquences protéiques a explosé. Afin d’acquérir une compréhension plus approfondie de la diversité et des fonctions des protéines, l’identification des protéines est particulièrement importante en biologie.
Dans le processus de reconnaissance des protéines, l’identification de l’homologie de séquence protéique est l’une des tâches les plus importantes.Cela peut aider les scientifiques à comprendre les relations évolutives, les caractéristiques structurelles et les fonctions des protéines. Bien que les méthodes traditionnelles d’alignement des séquences protéiques fonctionnent bien dans de nombreux cas, elles ne sont pas en mesure de gérer les homologues distants. Ces homologues distants sont souvent négligés dans les alignements de routine en raison de leur faible similarité de séquence, limitant ainsi la compréhension globale des chercheurs de la diversité et de la complexité des protéines.
Pour résoudre les problèmes de la recherche sur l'homologie des protéines à distance, basée sur des modèles de langage protéique et une technologie de récupération dense, Li Yu de l'Université chinoise de Hong Kong, en collaboration avec Sun Siqi, un jeune chercheur du Laboratoire des systèmes complexes intelligents de l'Université Fudan et du Laboratoire d'intelligence artificielle de Shanghai, et Mark Gerstein de l'Université de Yale, ont proposé un cadre de détection d'homologie ultra-rapide et très sensible - le Dense Homology Retriever (DHR).
DHR peut identifier des homologues distants cachés au plus profond de la séquence sans s'appuyer sur l'alignement de séquence traditionnel, grâce aux puissantes capacités de la structure du double encodeur et du modèle de langage protéique, apportant une vitesse et une sensibilité sans précédent à l'identification des homologues. La recherche a été publiée dans la revue de renommée internationale Nature Biotechnology sous le titre « Détection rapide et sensible d'homologues de protéines à l'aide d'une récupération dense et profonde ».
Points saillants de la recherche :
* Par rapport aux méthodes précédentes, le DHR améliore la sensibilité de plus de 10% et améliore la sensibilité de plus de 56% au niveau de la superfamille pour les échantillons difficiles à identifier à l'aide de méthodes basées sur l'alignement
* Le code DHR interroge les séquences et les bases de données 22 fois plus rapidement que les méthodes traditionnelles telles que PSI-BLAST et DIAMOND, et 28 700 fois plus rapidement que HMMER

Adresse du document :
https://doi.org/10.1038/s41587-024-02353-6
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Construire des ensembles de données dans plusieurs dimensions pour explorer un spectre plus large de séquences protéiques
L'ensemble de formation construit dans cette étude comprend 2 millions de séquences de requêtes soigneusement sélectionnées à partir de l'UR90.En utilisant l'algorithme JackHMMER, cette étude a recherché de manière itérative des séquences candidates dans Uni-Clust30 et a aligné les séquences candidates avec l'alignement de séquences multiples (MSA). Chaque MSA contenait 1 000 homologues, garantissant que seules les séquences les plus apparentées étaient conservées. Après un criblage rigoureux, JackHMMER a été redéployé pour traiter les différentes séquences obtenues et a utilisé les mêmes paramètres d'hyperparamètres que AF2 (AlphaFold 2) pour faciliter une comparaison équitable.
Dans l’étude de grands ensembles de données, l’étude a sélectionné l’ensemble de données BFD/MGnify.Il s’agit d’une base de données massive d’environ 300 millions de protéines, permettant l’exploration d’un spectre plus large de séquences protéiques.
La méthode DHR : un pipeline de recherche d'homologie protéique ultra-rapide et sensible
L'idée principale de la méthode DHR est de coder des séquences protéiques dans des vecteurs d'intégration denses afin de calculer efficacement la similarité entre les séquences.Plus précisément, cette étude a efficacement formé l'encodeur de séquence en initialisant l'ESM et en intégrant des techniques d'apprentissage contrastif, créant ainsi les conditions pour la construction d'un modèle de langage protéique et permettant à DHR d'être utilisé plus efficacement pour récupérer des homologues.
Comme le montre la figure a ci-dessous, une fois la phase de formation du double encodeur terminée, l’étude est en mesure de générer des séquences protéiques hors ligne de haute qualité. L’étude a ensuite utilisé ces intégrations et ces algorithmes de recherche de similarité pour récupérer des homologues pour chaque protéine de requête. En spécifiant la similarité comme mesure de récupération, les protéines similaires peuvent être trouvées plus précisément que les méthodes traditionnelles, et la similarité entre deux protéines peut être utilisée pour une analyse plus approfondie. Finalement, JackHMMER a construit le MSA des homologues récupérés, et l'étude a obtenu la technologie DHR qui peut découvrir rapidement et efficacement des homologues.
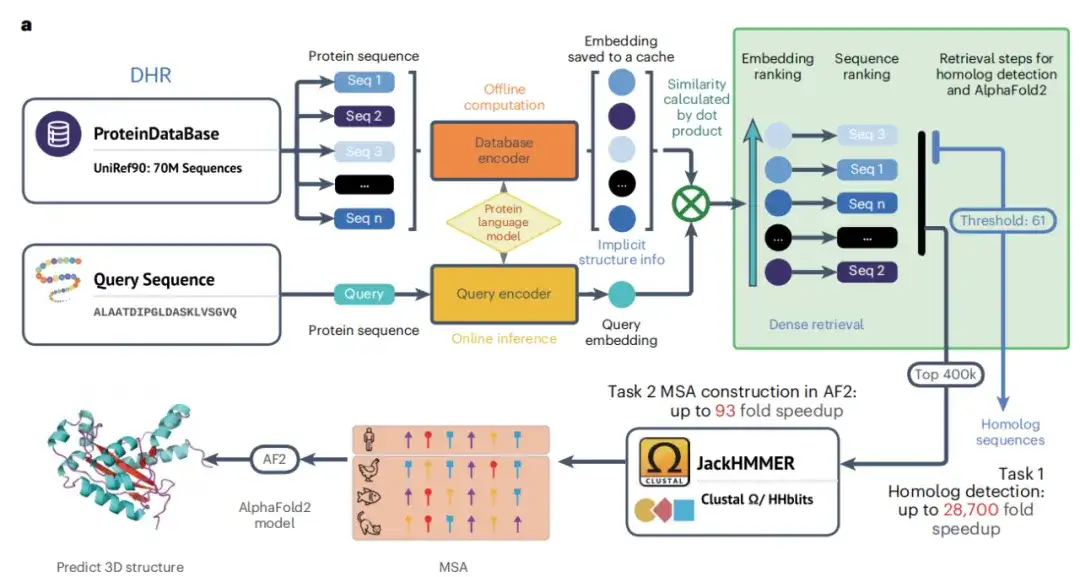
De plus, l'étude a également développé un modèle hybride DHR-meta, qui a surpassé les pipelines individuels sur les cibles CASP13DM (séquence de domaine) et CASP14DM en combinant DHR et AF2 par défaut.
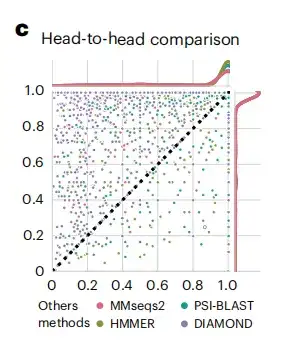
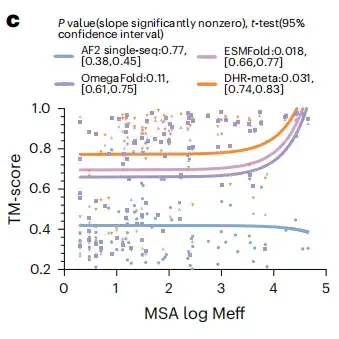
Après avoir obtenu les inclusions protéiques générées, l'étude a évalué les performances du DHR en le comparant aux méthodes de l'ensemble de données standard SCOPe (Structure Classification of Proteins).Comme le montre la figure c ci-dessous, la sensibilité des données DHR est meilleure que celle des autres méthodes.
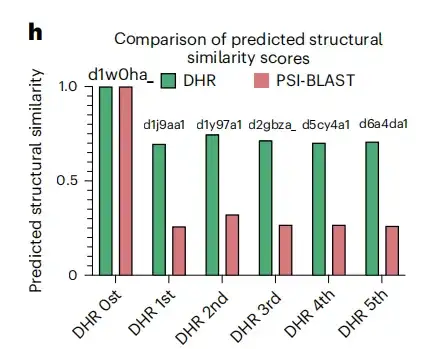
De plus, comme le montre la figure h ci-dessous, dans l'exemple spécifique de la requête d1w0ha, ni PSI-BLAST ni MMseqs2 n'ont correspondu à aucun résultat, mais DHR a récupéré 5 homologues, qui ont également été classés dans la même famille que d1w0ha dans SCOPe. Cela signifie que le DHR peut capturer davantage d’informations structurelles. Comparé aux méthodes traditionnelles telles que PSI-BLAST, MMseqs2, DIAMOND et HMMER, DHR a détecté le plus d'homologues (sensibilité de 93 %).Cela démontre que DHR est capable d’intégrer des informations structurelles riches et d’atteindre une sensibilité de 100 % dans de nombreux cas.
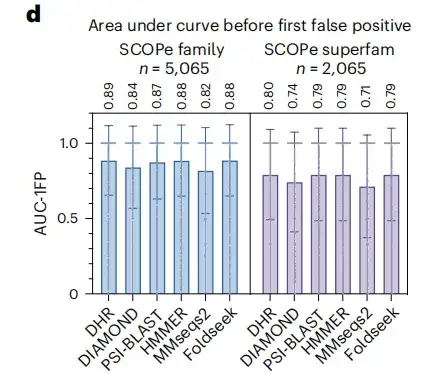
Pour renforcer la crédibilité des résultats, cette étude a également inclus un autre indicateur standard, l’aire sous la courbe avant le premier PF. Les résultats montrent que, comme le montre la figure d ci-dessous, DHR atteint un score de 89%.Parallèlement, d’autres méthodes ont également montré des performances comparables à celles de DHR, mais leur temps d’exécution était significativement plus long.Lorsque nous sommes passés au niveau de la superfamille pour analyser l’ensemble plus complexe des homologues distants, toutes les méthodes ont connu une baisse significative des performances, avec une baisse globale d’environ 10%. Malgré cela, DHR maintient toujours ses performances de premier plan avec un score AUC-1FP aussi élevé que 80%.
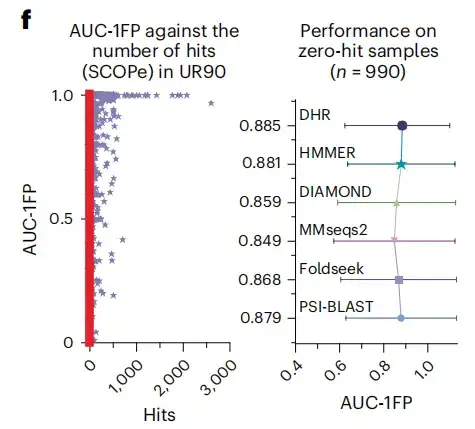
L'étude a également révélé que lors de l'utilisation de BLAST pour comparer la base de données SCOPe et UniRef90, la plupart des échantillons produisaient moins de 100 correspondances, et même environ 500 échantillons n'obtenaient aucune correspondance, indiquant que ces échantillons étaient des structures « invisibles » dans l'ensemble de données d'entraînement. En revanche, le DHR parvient toujours à des prédictions de haute qualité pour ces structures, atteignant un score AUC-1FP de 89%.Cela démontre la capacité du DHR à gérer des données entièrement nouvelles.
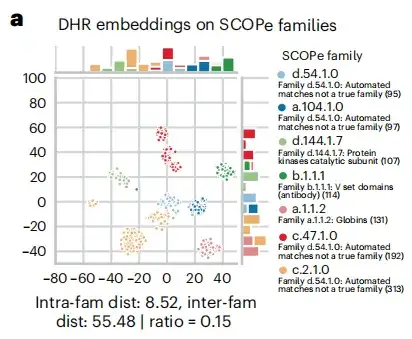
Au cours du processus de recherche d'homologie, comme le montre la figure a ci-dessous, l'étude a révélé que l'intégration de séquences DHR contient une grande quantité d'informations structurelles et que la précision de DHR dans la récupération de substances homologues dépasse même celle des méthodes d'alignement basées sur la structure. Sur la base de ce résultat,Cette étude a également révélé la corrélation entre le classement de similarité de séquence et la similarité structurelle du DHR.
Résultats de recherche : DHR offre une meilleure précision et une meilleure efficacité, et peut créer des MSA de haute qualité sur des ensembles de données à grande échelle
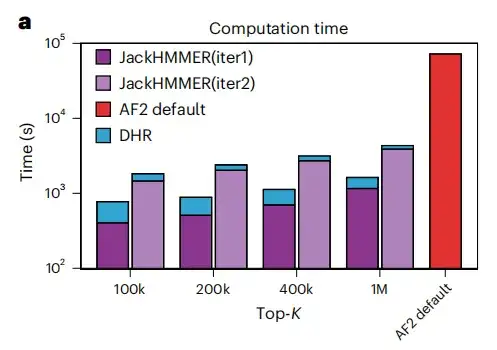
Nous avons créé des MSA à partir de JackHMMER en utilisant des homologues fournis par DHR et les avons comparés avec le pipeline par défaut AF2. Comme le montre la figure a ci-dessous, la vitesse de fonctionnement moyenne de toutes les configurations de DHR + JackHMMER est plus rapide que celle du JackHMMER ordinaire d'AF2. De plus, DHR chevauche JackHMMER d'environ 80% lors de la construction de MSA sur UniRef90.Cela suggère que de nombreuses tâches en aval liées à MSA peuvent être effectuées à l'aide de DHR, produisant des résultats similaires mais plus rapidement.
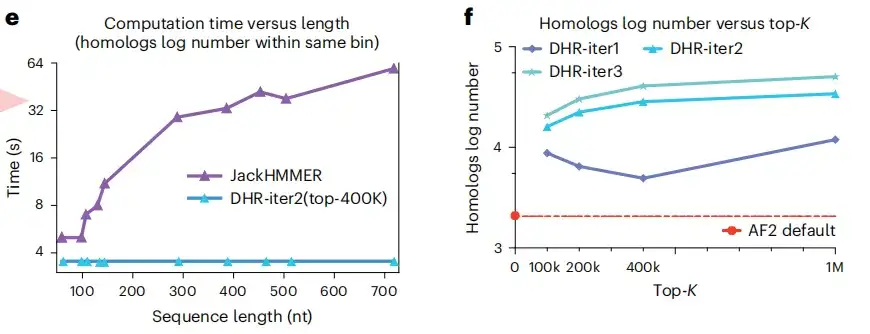
Comme le montrent les figures e et f ci-dessous, un autre avantage du DHR est qu'il peut construire le même nombre d'homologues de longueurs différentes dans un temps constant, tandis que JackHMMER évolue linéairement. De plus, par rapport à AF2, DHR peut fournir plus d'homologues et de MSA pour l'intégration des requêtes. Ces résultats indiquent queLe DHR est une approche prometteuse pour toutes les catégories de construction MSA.
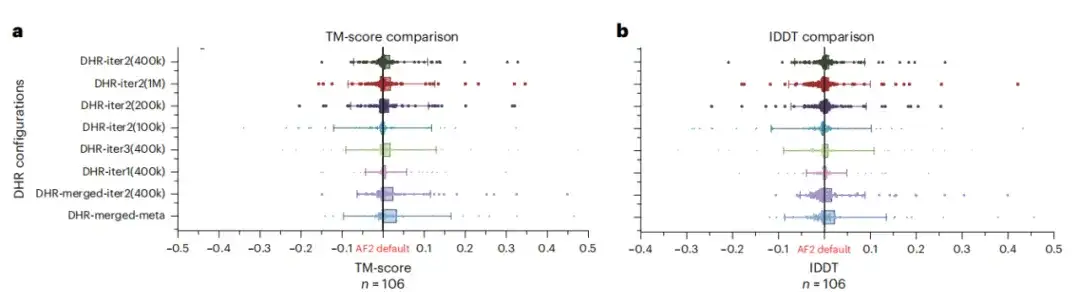
Bien que le DHR soit capable de produire différents MSA, cette étude a analysé plus en détail s'il pouvait servir de complément au MSA de base de l'AF2. Les résultats de la recherche montrent que, comme le montrent les figures a et b ci-dessous, les performances de la combinaison de tous les MSA et AF2 sous différents paramètres DHR sont les meilleures.Cela signifie que DHR peut réapprovisionner rapidement et avec précision le pipeline MSA d'AF2.
Afin d’examiner les avantages potentiels des grands modèles de langage pour la prédiction de la structure des protéines, cette étude a évalué si le remplacement de MSA par un grand modèle de langage sur toutes les cibles CASP14DM produirait de meilleurs résultats. Comme le montre la figure c ci-dessous, dans le cas simple avec un grand nombre d’AMS disponibles, le modèle de langage peut transmettre autant d’informations que les AMS. Cependant, à mesure que la longueur de la séquence augmente, les performances de DHR-meta s'améliorent de plus en plus, surpassant ESMFold dans presque tous les cas. Cela signifie que par rapport aux méthodes basées sur des modèles de langage,Le modèle basé sur MSA peut considérablement améliorer la précision et l’efficacité des prédictions.
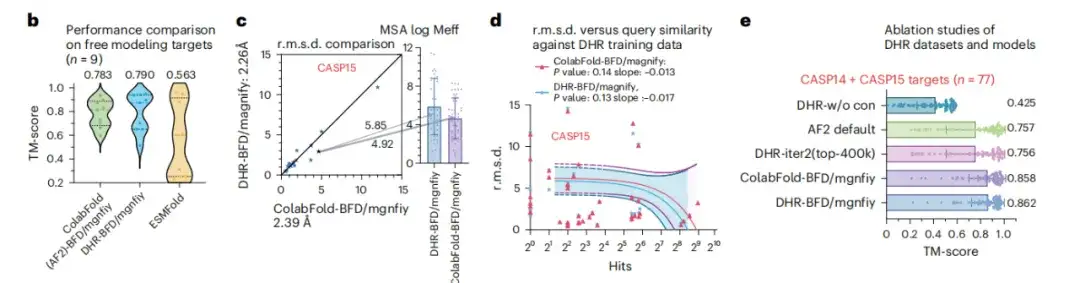
Afin d'étudier l'évolutivité du DHR dans de grands ensembles de données, cette étude a mené une analyse approfondie du DHR basée sur BFM/MGnify. Comme le montre la figure b ci-dessous, dans le scénario complexe de prédiction de la structure des cibles FM, DHR est capable de se démarquer en générant des MSA plus significatifs, avec la méthode ColabFold utilisant MMseqs2 pour construire des MSA surpassant de 0,007 score TM.
Dans la figure 2c, DHR montre une légère amélioration des performances par rapport à ColabFold-MMseqs2. La figure d ci-dessous montre également qu'après les tests de similarité de CASP14 et SCOPe, il a été constaté que DHR ne se souvenait pas simplement des résultats de la requête ou du hit, mais effectuait une évaluation complète de la similarité de toutes les cibles. Ces résultats prouvent queDHR permet la construction de MSA de protéines désordonnées sur des ensembles de données de recherche à grande échelle avec une grande diversité.
Jeunes forces dans le domaine de la prédiction de la structure des protéines
Il ne fait aucun doute que la prédiction de la structure des protéines joue un rôle important dans des applications telles que le développement de médicaments et la conception d’anticorps. L’IA pourrait devenir la clé pour résoudre le problème historique de la précision limitée de la prédiction de la structure des protéines. Dans ce domaine clé, les équipes de recherche scientifique nationales ont progressivement formé une tendance d’une centaine d’écoles de pensée, et les jeunes chercheurs émergents sont devenus une force incontournable. Li Yu et Sun Siqi, qui ont dirigé les résultats de recherche ci-dessus, sont tous deux parmi les meilleurs.

Li Yu a obtenu sa licence (avec mention) en sciences biologiques de la classe d'élite Bei Shizhang de l'Université des sciences et technologies de Chine en 2015, sa maîtrise en informatique de l'Université des sciences et technologies du roi Abdallah (KAUST) en Arabie saoudite en décembre 2016, et son doctorat en informatique de la même université en 2020.
En décembre de la même année, il retourne en Chine et rejoint le département d'informatique et d'ingénierie de l'Université chinoise de Hong Kong en tant que professeur adjoint, dirigeant le groupe Intelligence artificielle dans les soins de santé (AIH). Il a mené des recherches approfondies à l’intersection de l’apprentissage automatique, des soins de santé et de la bioinformatique, et a dirigé l’équipe pour développer de nouvelles méthodes d’apprentissage automatique pour résoudre les problèmes informatiques en biologie et en soins de santé, en particulier les problèmes d’apprentissage structuré.
Concernant les domaines de la biologie et des soins de santé dans lesquels il a été profondément impliqué, Li Yu a déclaré : « Mon objectif à long terme est d'améliorer le système de santé et de bénéficier directement à la société en améliorant la santé et le bien-être des gens. »Il convient de mentionner qu'il a également été sélectionné dans la liste Forbes Asia « 30 Under 30 » 2022 (Santé et Sciences).

Sun Siqi a obtenu d'excellents résultats au concours mondial de prédiction de la structure des protéines et est actuellement un jeune chercheur au Laboratoire de théorie fondamentale et de technologie clé des systèmes complexes intelligents et au Laboratoire d'intelligence artificielle de Shanghai de l'Université Fudan.Il s'engage dans la recherche d'application de l'apprentissage profond dans des domaines interdisciplinaires tels que les sciences de la vie et le traitement du langage naturel, et se concentre sur l'amélioration de la précision et de la vitesse des modèles et sur la résolution de problèmes spécifiques dans la mise en œuvre des modèles.
En termes de prédiction des protéines, il se concentre sur la prédiction de la structure et de la séquence des protéines grâce à des modèles d'apprentissage profond et à des modèles de formation pour identifier les modèles et les régularités dans les séquences afin de prédire la séquence et le repliement des protéines, améliorant ainsi la précision et l'efficacité du séquençage de novo des protéines et de la prédiction de la structure, et créant de nouvelles possibilités pour la conception de médicaments et le traitement des maladies.
Dans le domaine national de l’IA4S, de plus en plus de jeunes forces sont actives. On peut prévoir que la technologie de l’IA jouera un rôle plus critique dans le domaine de la prédiction de la structure des protéines, mais le chemin à parcourir est long et ardu. Il est gratifiant de constater que l’équipe de recherche scientifique nationale a fait preuve d’un esprit tenace d’exploration et d’une capacité d’innovation. Ils ont non seulement travaillé dur sur l’optimisation des algorithmes et la construction de modèles, mais ont également mené des recherches approfondies sur le traitement des données, la vérification expérimentale, etc. pour garantir la scientificité et la praticité des résultats de la recherche. Ces efforts se transforment progressivement en applications pratiques, apportant une nouvelle vitalité et un nouvel espoir à des domaines tels que la recherche et le développement médicaux et la biotechnologie.
Enfin, je vous recommande une activité de partage académique !
La troisième diffusion en direct de Meet AI4S a invité Zhou Ziyi, chercheur postdoctoral à l'Institut des sciences naturelles de l'Université Jiao Tong de Shanghai et au Centre national de mathématiques appliquées de Shanghai. Cliquez ici pour prendre rendez-vous pour regarder la diffusion en direct !
https://hdxu.cn/6Bjomhdxu.cn/6Bjom