Command Palette
Search for a command to run...
Démontage Complet d'AlphaFold 3, Zhong Bozitao De l'Université Jiaotong De Shanghai : Utilisation Extrême Des Données Pour Prédire Toutes Les Structures Biomoléculaires Avec Une Précision Atomique, Mais Ce n'est Pas Parfait

AlphaFold 3, capable de prédire les structures et les interactions de toutes les biomolécules avec une « précision atomique », a suscité de nombreuses discussions dans l'industrie dès sa sortie. Le 13 août, à l'école d'été d'IA pour la bio-ingénierie de l'Université Jiao Tong de Shanghai,Le Dr Zhong Bozitao a systématiquement trié son expérience d'apprentissage sur le thème « AlphaFold 3 : Principes, Applications et Perspectives », et a trié de manière approfondie de nombreux résultats de recherche pertinents de la communauté de recherche scientifique, partageant ses connaissances approfondies sur AlphaFold 3 avec tout le monde.HyperAI a organisé le contenu principal du discours sans violer l’intention initiale. Voici la transcription du discours.

En nous concentrant sur la prédiction de la structure des protéines, nous parlerons aujourd'hui d'AlphaFold 3, qui est actuellement le meilleur outil de prédiction de la structure moléculaire biologique des protéines et encore plus étendu.Le statut d’AlphaFold 3 est évident.
La synthèse des protéines commence par la transcription de l'ADN, qui transfère ensuite l'information génétique à l'ARN, qui est ensuite traduit en protéine et replié en structures secondaires, tertiaires et quaternaires. La plupart des protéines se replient dans des conformations uniques, et les informations nécessaires à la structure sont codées dans la séquence d'acides aminés, ce qui est ce que nous disons souvent : la séquence détermine la structure, et la structure détermine la fonction.La prédiction de la structure des protéines est essentielle à la compréhension des fonctions biologiques.
Percée d'AlphaFold 3 : architecture de modèle innovante et utilisation améliorée des données
Comparaison des architectures des modèles AlphaFold 3 et AlphaFold 2
Dans le passé, AlphaFold 2 a directement « battu » d’autres algorithmes dans la prédiction de la structure des protéines.Son architecture de base peut être résumée en trois parties clés, comme le montre la figure ci-dessous : La première partie, le module MSA & Template dans la boîte bleue, est responsable de la collecte et de l'intégration des informations sur l'alignement de séquences multiples (MSA) et la structure du modèle comme données d'entrée pour le modèle. La deuxième partie, le module Evoformer dans la case verte, est responsable de la compréhension des informations co-évolutives dans les organisations multi-séquences, du raffinement et du traitement des informations collectées et de leur transmission au module Structure dans la case violette de la troisième partie.
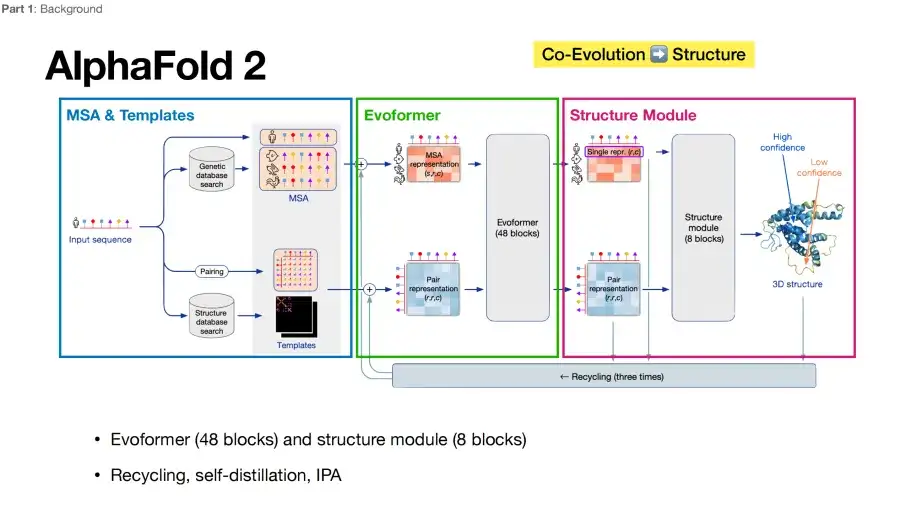
D'un point de vue d'apprentissage profond, Evoformer joue le rôle d'un encodeur, tandis que Structure Module équivaut à un décodeur.AlphaFold 2 a reçu de nombreux éloges en grande partie grâce à ses capacités d'optimisation de bout en bout, mappant directement de l'entrée de séquence à la sortie structurelle.
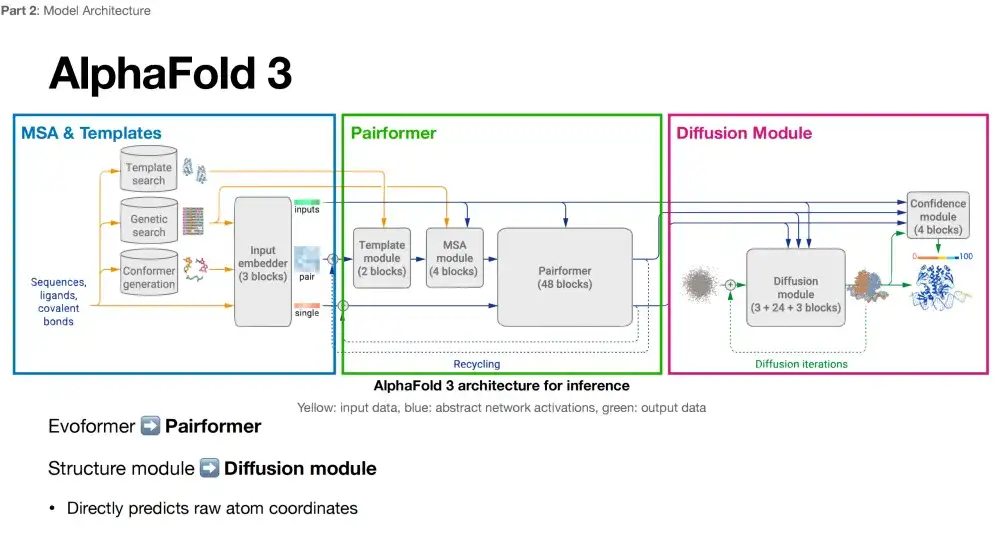
On pense généralement que les changements dans l’architecture du modèle d’AlphaFold 3 ne sont pas aussi importants qu’on l’imaginait. Son cadre modèle est également composé de 3 parties clés. La comparaison entre chaque partie et AlphaFold 2 est la suivante :
Partie 1 : Maintenir une grande similarité
Comme le montre la figure ci-dessous, en comparant les diagrammes d'architecture d'AlphaFold 3 et d'AlphaFold 2, on peut voir que la première partie d'AlphaFold 3 (dans la case bleue) inclut toujours MSA et Template, et introduit en plus le lien de génération Conformer.
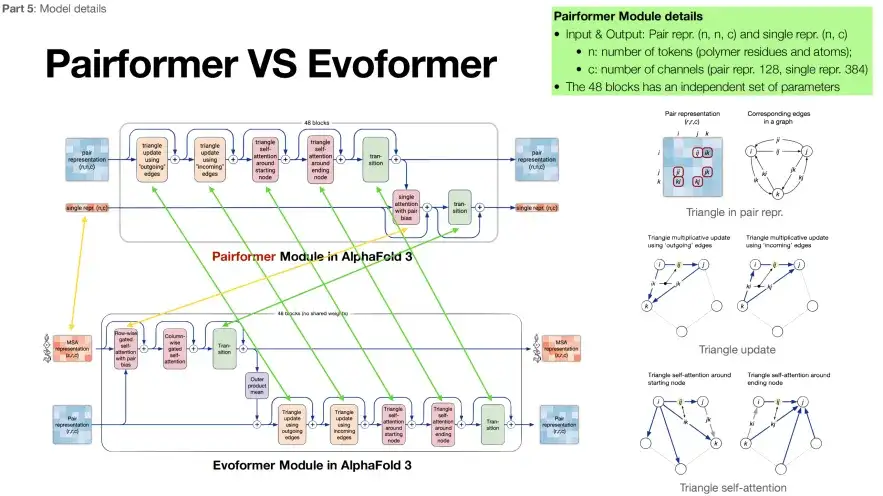
Partie 2 : Réduire la dépendance aux séquences MSA
La deuxième partie d'AlphaFold 3 (dans la case verte) s'appelle Pairformer. Sa structure est essentiellement très similaire à celle d'Evoformer, mais le nombre de modules MSA est réduit à 4. Comme le montre la figure ci-dessous, les flèches vertes indiquent le même contenu dans les deux modules et les flèches jaunes indiquent les différences. Comme vous pouvez le voir,AlphaFold 3 met davantage l’accent sur la séquence de la protéine cible et s’appuie moins sur la séquence MSA.
De plus, nous pensons que la raison pour laquelle AlphaFold 3 peut montrer de bonnes performances dans plusieurs tâches peut être qu'il réduit sa dépendance à l'alignement de séquences multiples (MSA).Comme le montre la figure ci-dessous, le côté droit montre l'impact du MSA sur les performances d'AlphaFold 2 : à mesure que le nombre de MSA augmente, après avoir dépassé un certain seuil (ligne rose), l'amélioration des performances d'AlphaFold 2 a tendance à se stabiliser. Comme on peut le voir dans la partie centrale de la figure ci-dessous, par rapport à AlphaFold 2, l’impact du MSA sur AlphaFold 3 est affaibli (la courbe fluctue très peu).
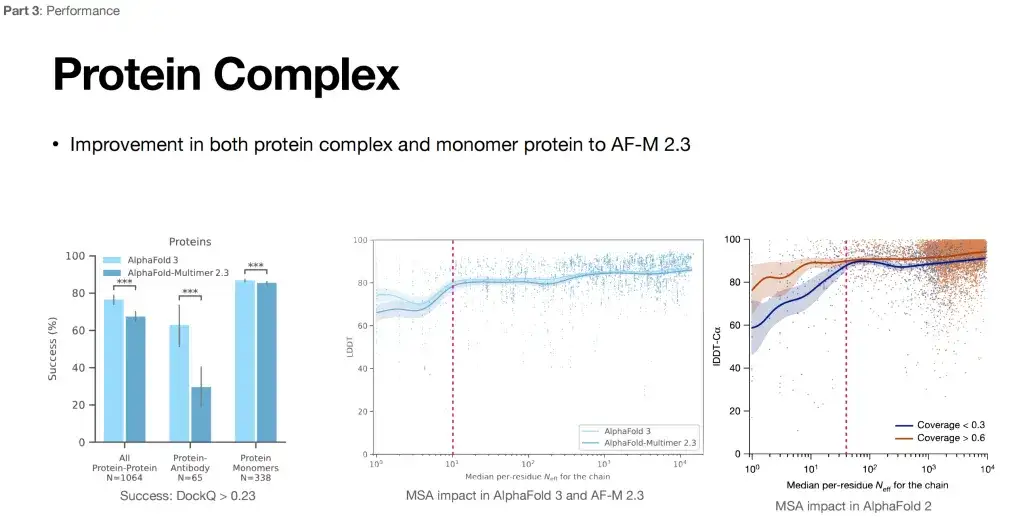
De plus, la maturation des anticorps nécessite souvent un processus d’hypermutation in vivo. Les informations MSA sont d’une aide limitée pour prédire leur structure, et il est difficile de trouver des informations MSA appariées pour les protéines et leurs complexes. De ce point de vue, l’élargissement du champ d’application d’AlphaFold 3 pourrait être la réduction de sa dépendance à l’égard du MSA.
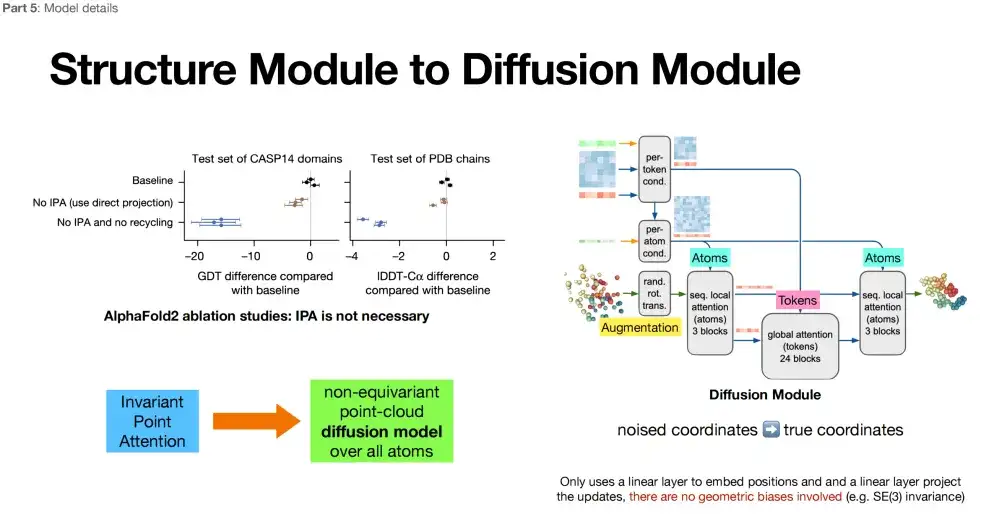
Partie III : Génération de structures entièrement atomiques + suppression de l'invariance de rotation stéréo
La troisième partie d'AlphaFold 3 (dans la case violette) adopte le modèle Diffusion, qui appartient également à la catégorie des modules de structure. La différence est que le modèle de diffusion remplace l’optimisation itérative répétée dans le module de structure par un nouveau mécanisme appelé modèle de diffusion.
* Modèle de diffusion : ajoutez du bruit au modèle (vers l'avant), laissez le modèle débruiter (vers l'arrière), apprenez le processus inverse et générez une distribution de données similaire.
Comme le montre la figure ci-dessous, dans la troisième partie, AlphaFold 3 parvient à générer une structure au niveau entièrement atomique. Les atomes, en tant qu'éléments de base des molécules, peuvent contenir des informations physiques plus riches, ce qui signifie qu'AlphaFold 3 peut capturer des lois physiques plus profondes lors de la prédiction des structures des protéines. De plus, AlphaFold 3 abandonne l'invariance de rotation stéréo soulignée dans AlphaFold 2. Après avoir supprimé l'architecture supplémentaire de cette fonctionnalité dans AlphaFold 2, les chercheurs ont constaté que la conception du modèle (module de diffusion) est devenue plus libre.
AlphaFold 3 améliore l'utilisation des données
Les ressources en données sur les protéines sont limitées, mais AlphaFold 3 non seulement élargit l'ensemble de données, mais améliore également leur utilisation.Plus précisément, comparé à l'ensemble de données de niveau million d'AlphaFold 2, AlphaFold 3 se rapproche directement du niveau milliard et l'ensemble d'entraînement est plus grand. De plus, en plus des données de la PDB, son ensemble d’entraînement intègre également une grande quantité d’autres données. Par exemple, les données structurelles prédites par AlphaFold 2 sont plus précises en tant qu’extension de l’ensemble d’entraînement. L'ensemble d'entraînement spécifique est présenté dans la figure ci-dessous :

AlphaFold 3 réalise un énorme bond en avant dans le champ d'application
Le plus grand changement d’AlphaFold 3 est qu’il a réalisé un saut qualitatif dans son champ d’application.Dans le passé, AlphaFold 2 était davantage axé sur la prédiction des structures d’acides aminés, tandis qu’AlphaFold 3 peut prédire directement les structures au niveau atomique. Son expansion fonctionnelle se reflète spécifiquement dans les quatre aspects suivants :
* Capable de prédire avec précision les ligands, c'est-à-dire de prédire les sites de liaison des petites molécules dans les protéines ;
* Capable de prédire la structure d’un complexe protéique ;
* Capable de prédire les structures de modification post-traductionnelle des protéines et des acides nucléiques ;
* Capable de prédire les structures de l’ADN et de l’ARN, ainsi que les structures des complexes d’ADN/ARN et des protéines.
AlphaFold 3 change le domaine de l'amarrage des ligands
Parmi eux, l’impact le plus important d’AlphaFold 3 sur le domaine scientifique est l’amélioration de la tâche d’amarrage des ligands.Comme le montre la figure ci-dessous, les taux de réussite de différents algorithmes d'apprentissage en profondeur dans 4 tâches d'amarrage de ligand différentes sont évalués dans le cadre du benchmark PostBusters. On peut constater qu'AlphaFold 3 peut atteindre le taux de réussite le plus élevé, c'est-à-dire 76,4%, sur la base de poches inconnues et de connaissances structurelles préalables.
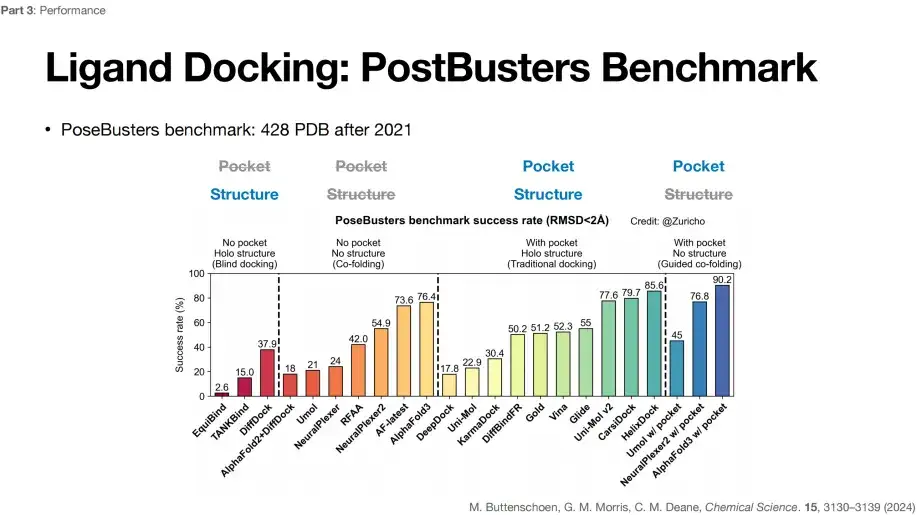
PostBusters Benchmark sélectionne 428 données PDB de 2021 et après
La norme de réussite de la tâche est que l'écart entre la position d'amarrage prévue de la petite molécule et la position d'amarrage réelle soit inférieur à 2 Å.
Comme le montre la figure ci-dessus, dans le premier type de tâche d'amarrage aveugle, avec une position de poche inconnue et une structure protéique connue (pas de poche, structure Holo), DiffDock peut atteindre le taux de réussite le plus élevé de 37,9%.
Dans le deuxième type de tâche de co-pliage (les petites molécules et les structures protéiques sont pliées), avec des positions de poche inconnues et des structures protéiques inconnues (pas de poche, pas de structure), le taux de réussite de la prédiction combinée AlphaFold 2+DiffDock est tombé à 18%,De plus, AlphaFold 3 a obtenu le taux de réussite le plus élevé de 76,4%, ce qui montre qu'AlphaFold 3 non seulement prédit avec précision, mais ne s'appuie pas non plus sur une connaissance préalable des poches et des structures.
Dans la troisième catégorie de tâches d'amarrage traditionnelles, la position de la poche de la petite molécule et la structure de la protéine sont connues (avec poche, structure Holo), c'est-à-dire que la poche est exposée. Gold a atteint un taux de réussite de 51,2%, Vina a montré un taux de réussite de 52,3% et Glide a augmenté à 55%. D’autres algorithmes d’apprentissage en profondeur peuvent également atteindre des niveaux relativement bons, ce qui indique que le taux de réussite est affecté par la poche.
Dans la quatrième catégorie de tâches de co-pliage guidé, avec des positions de poche connues et des structures protéiques inconnues (avec poche, sans structure), le taux de réussite du modèle a été considérablement amélioré, de 76,4% à 90,2% pour AlphaFold 3, indiquant que les informations de poche connues peuvent améliorer le taux de réussite de la tâche. Cependant, il existe une certaine controverse sur la définition de poche.Par conséquent, si vous souhaitez connaître l'amélioration spécifique d'AlphaFold 3 sur la tâche d'amarrage du ligand, vous ne pouvez considérer que le taux de réussite du deuxième type de tâche, qui est relativement plus stable.
Comme le montre la figure ci-dessous, il existe des différences significatives dans les définitions de poche entre les différents modèles. La poche d'or est une sphère de 25 Å (la partie bleue dans le coin supérieur gauche de la figure), tandis que le modèle Vina utilise un cube de 25 Å comme représentation de poche, la taille de la poche DeepDock est de 10 Å et la taille de la poche Uni-Mol est de 8 Å.
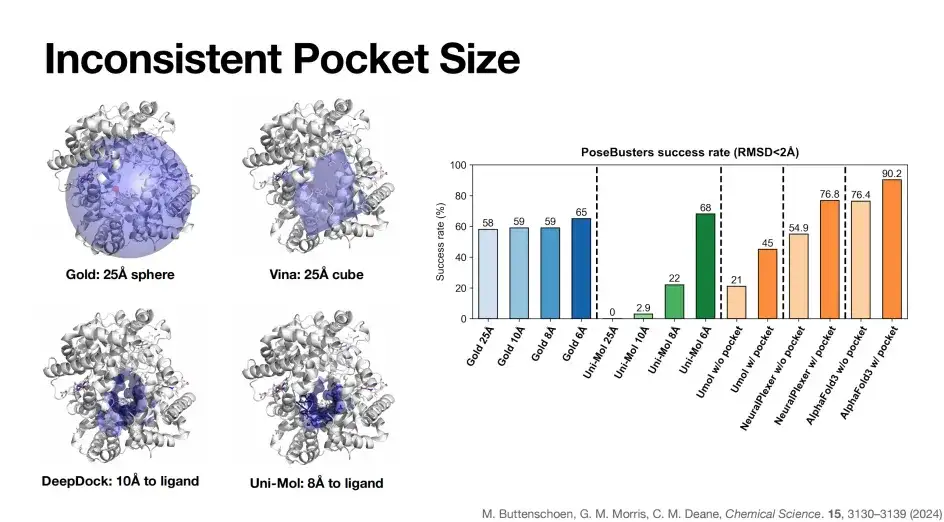
Comme le montre le côté droit de la figure ci-dessus, lorsque la taille de la poche du modèle Gold est progressivement réduite de 25 Å à 6 Å, son taux de réussite de référence PoseBusters est relativement stable, ce qui est dû aux caractéristiques de l'algorithme basé sur la physique de Gold. En revanche, l'algorithme d'apprentissage profond Uni-Mol a progressivement réduit la poche à 6 Å, le taux de réussite augmentant jusqu'à 68%, puis chutant à zéro à 25 Å, reflétant la dépendance de certains algorithmes d'amarrage d'apprentissage profond sur la poche.
De même, comme mentionné précédemment, après l’introduction des informations de poche, le taux de réussite d’amarrage d’AlphaFold 3 a été considérablement amélioré, passant de 76,4% à 90,2%.En résumé, les informations de poche jouent un rôle clé dans l’amélioration du taux de réussite de la prédiction du modèle. Mais idéalement, un modèle capable d’atteindre une grande précision sans informations de poche ou structurelles serait notre meilleur choix, comme AlphaFold 3.
AlphaFold 3 permet la prédiction de la structure des anticorps et des antigènes
Une autre application d’AlphaFold 3 est la prédiction de la structure des anticorps et des antigènes. Sur le côté gauche de la figure ci-dessous se trouve l’évaluation des performances d’AlphaFold 3 pour la prédiction de la structure des anticorps et des antigènes. Sous une norme d'évaluation inférieure (DockQ>0,23), avec une seule exécution, le taux de réussite d'AlphaFold 3 est inférieur à 40% (ligne bleu clair), mais après 1 000 tentatives, le taux de réussite de prédiction peut être amélioré à 60%.
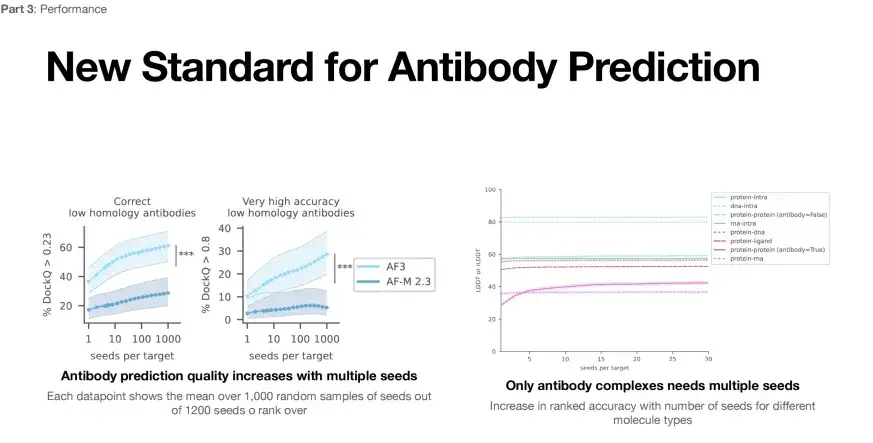
* Gauche : Prédiction de la structure de l'anticorps, chaque point de données représente le score moyen de 1 000 graines sélectionnées au hasard parmi 1 200 graines
* Droite : Lorsque l’indice d’évaluation DockQ est supérieur à 0,23, on peut considérer que la précision structurelle reste à vérifier ; lorsque DockQ dépasse 0,8, la prédiction structurelle est très précise
De plus, si l'on mesure selon une norme plus stricte (DockQ>0,8), le taux de réussite d'une seule exécution peut être aussi bas que 10%, et en augmentant le nombre d'exécutions à 1 000, le taux de réussite peut être augmenté à 30%.Cela montre que nous pouvons améliorer le taux de réussite de la prédiction de la structure de l'antigène de l'anticorps en augmentant le nombre d'exécutions AlphaFold 3 (graines par cible).
Cependant, comme le montre le côté droit de la figure ci-dessus, AlphaFold 3 ne peut améliorer le taux de réussite qu'en augmentant le nombre d'exécutions lors de la prédiction de la structure des complexes protéine-protéine. Cela montre que l’applicabilité d’AlphaFold 3 pour prédire d’autres types de structures complexes doit également être optimisée davantage.
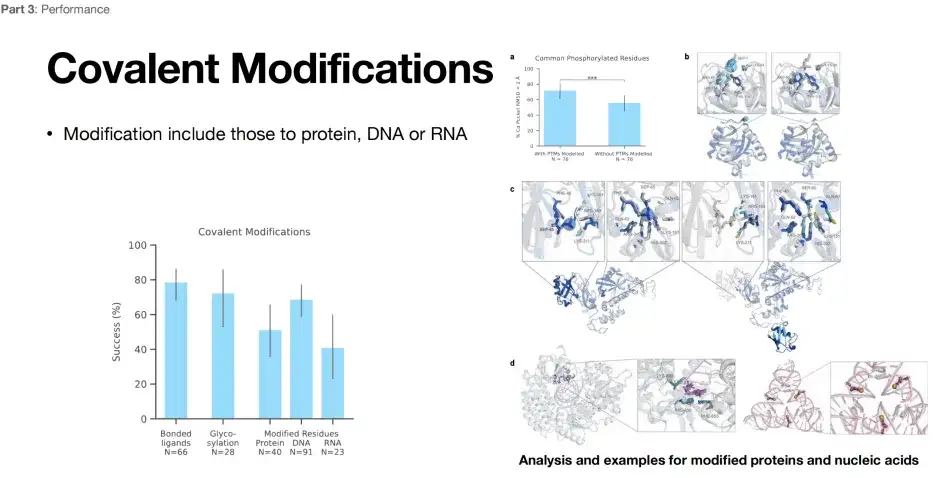
AlphaFold 3 permet la prédiction des modifications covalentes
Comme le montre la figure ci-dessous, AlphaFold 3 démontre également d’excellentes capacités de prédiction structurelle en termes de prédiction de modification.Le taux de réussite peut atteindre environ 80%, 60% et 40%. AlphaFold 3 est sans aucun doute un outil puissant pour les chercheurs engagés dans les modifications covalentes.
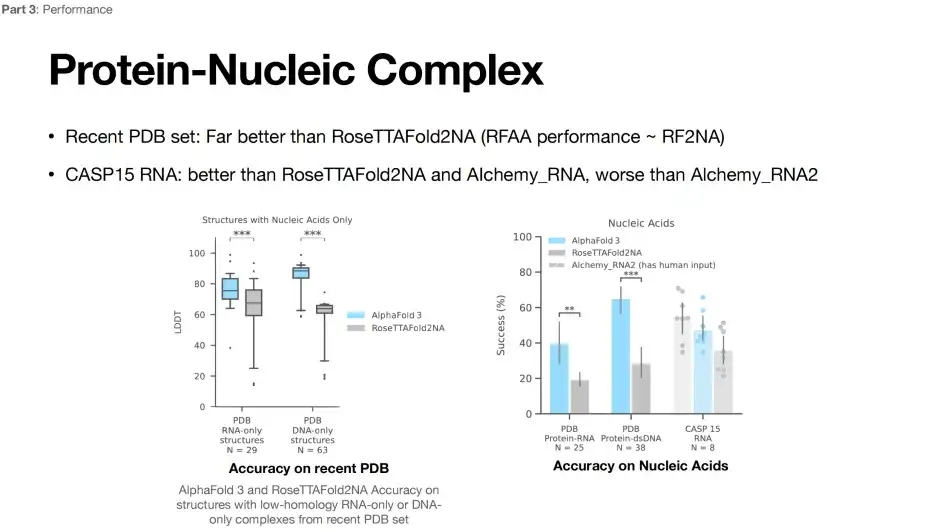
Limites d'AlphaFold 3 dans la prédiction de la structure de l'ARN
Actuellement, la prédiction de la structure de l’ARN est encore difficile.Comme le montre la figure ci-dessous, AlphaFold 3 a considérablement amélioré les performances de prédiction par rapport au modèle RoseTTAFoId2NA. Cependant, lors de la prédiction de la structure de l'ARN CASP15, la précision d'AlphaFold 3 est inférieure à celle du modèle Alchemy_RNA2 (avec apport humain).
Comparaison des avantages et des inconvénients d'AlphaFold 3 sur différentes tâches
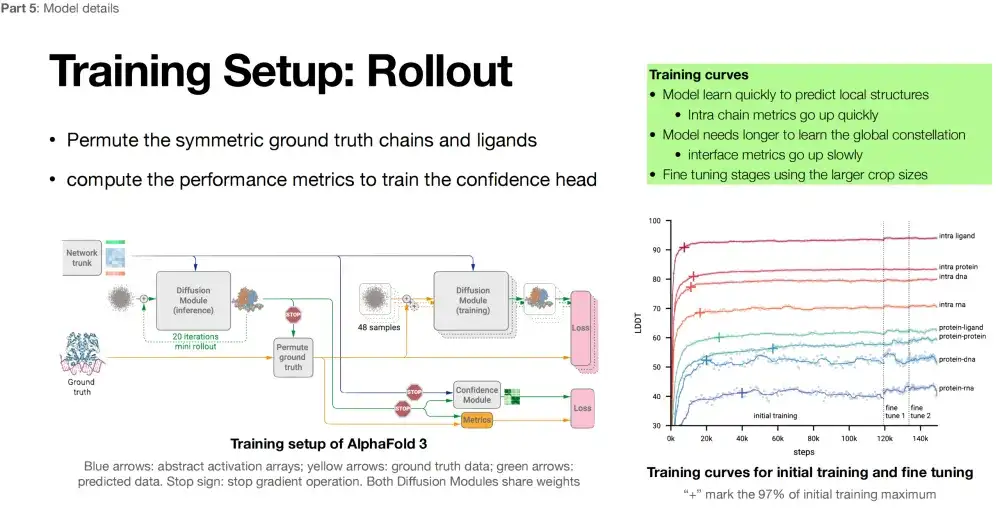
En analysant la courbe d’entraînement d’AlphaFold 3, nous pouvons clairement voir les performances du modèle sur différentes tâches. Plus l'indicateur LDDT est élevé, mieux c'est.Comme le montre la figure ci-dessous, le modèle est le plus performant pour prédire les structures intra-ligands ; il montre également une grande précision dans la prédiction des structures intra-protéiques ; le modèle est également performant dans la prédiction des structures intra-ADN, grâce à la structure stable en double hélice de l'ADN ; en revanche, le modèle est peu performant dans la prédiction des structures intra-ARN.
En ce qui concerne le domaine de la prédiction complexe, le modèle a obtenu les meilleurs résultats dans la prédiction de la structure du complexe protéine-ligand, suivi de la prédiction du complexe protéine-protéine. Dans la prédiction du complexe protéine-ADN, les performances du modèle ont diminué et la prédiction du complexe protéine-ARN a obtenu les pires résultats. Ce résultat reflète également la difficulté de prédire la structure de l’ARN. Les données structurelles de l’ARN sont rares et la structure est dynamique et flexible, ce qui constitue l’un des défis actuels rencontrés dans le domaine de la biologie structurale.
De plus, lorsque les chercheurs ont utilisé AlphaFold 3 pour la prédiction de la structure,La fiabilité des résultats de prédiction peut également être évaluée via le tableau PAE.

AlphaFold 3 n’est pas parfait
AlphaFold 3 n'est pas parfait. Par exemple, il peut trouver la mauvaise chiralité.Si une situation anormale survient pendant l'opération, il est recommandé d'exécuter le test plusieurs fois pour vérifier la stabilité des résultats. Deuxièmement, AlphaFold 3 présente également des limites dans la prédiction de la dynamique des protéines, ce qui peut être dû au manque de données structurelles et à l’incapacité de saisir les informations conformationnelles multidimensionnelles des protéines.
*Si un objet est différent de son image miroir, il est dit « chiral » et son image miroir ne peut pas être superposée à l'objet d'origine, tout comme les mains gauche et droite sont des images miroir l'une de l'autre et ne peuvent pas être superposées.
De plus, AlphaFold 3 présente également un problème commun aux modèles génératifs, à savoir l’hallucination.Comme le montrent les résultats de prédiction de la structure des protéines ci-dessous, seule la zone grise de la structure des protéines à gauche peut être résolue, et le reste peut être dans un état déplié en raison d'une densité électronique insuffisante. L'image du milieu est le résultat de la prédiction de la protéine par AlphaFold 2. La zone bleue est considérée comme étant pliée et les autres parties du « ruban » sont considérées comme dépliées. La structure prévue est relativement raisonnable. À droite se trouve le résultat de prédiction d'AlphaFold 3, qui tend à plier toutes les zones pliables possibles. Cette structure semble raisonnable, mais en réalité, la plupart des zones ci-dessus ne sont pas réellement pliées.Par conséquent, l’illusion d’AlphaFold 3 tend à prédire que les protéines sont pliées plutôt que de conserver leur éventuel état déplié.

Pour résoudre le problème d'hallucination d'AlphaFold 3,Les chercheurs ont choisi une méthode directe et efficace : puisque les résultats prédits par AlphaFold 2 sont relativement raisonnables, les résultats prédits par AlphaFold 2 seront intégrés dans l'ensemble de données d'entraînement d'AlphaFold 3 pour améliorer l'effet d'entraînement du modèle. Cependant, cette méthode présente une limitation : s’il y a des erreurs dans la prédiction d’AlphaFold 2 elle-même, cela peut affecter la qualité de prédiction d’AlphaFold 3, à moins que d’autres sources de données puissent être introduites pour optimiser davantage le modèle.
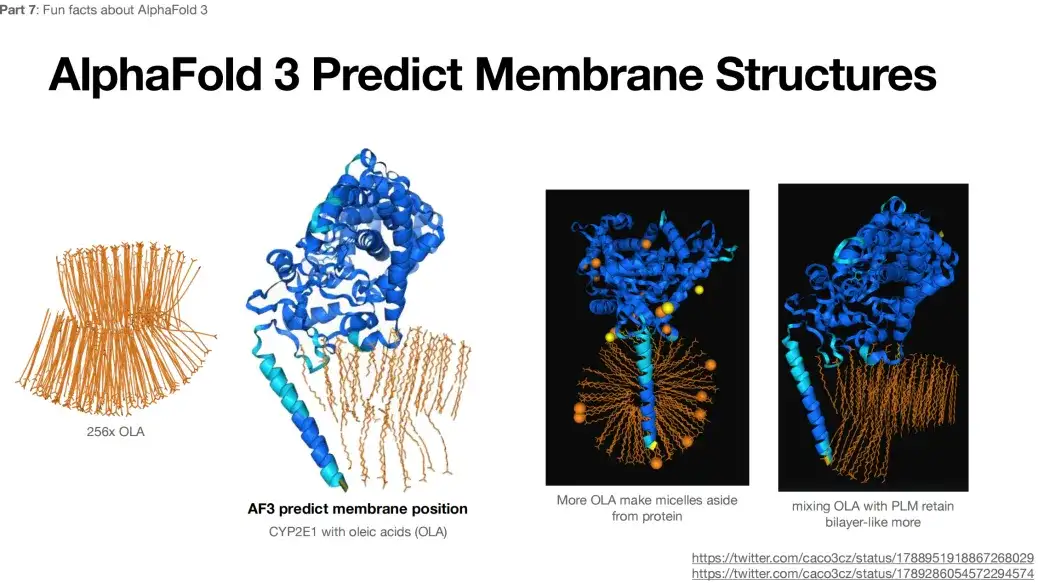
De plus, lorsque 256x OLA est soumis en entrée à AlphaFold 3, le résultat prédit montre une structure de type bicouche, comme illustré dans la figure ci-dessous.Cette structure n’est pas attendue ou typique.
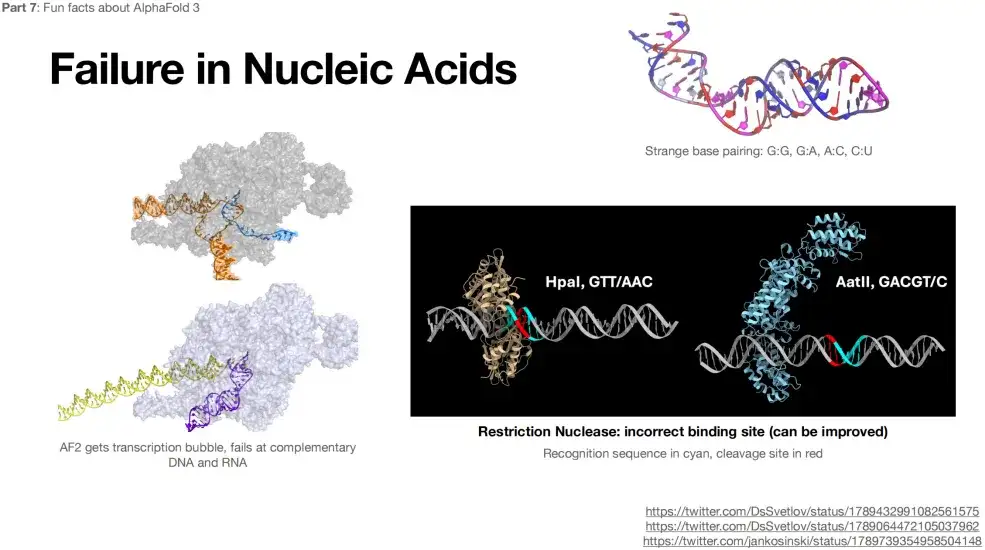
De plus, AlphaFold 3 n’est pas précis dans la prédiction des structures de l’ARN et de l’ADN.Comme le montre la figure ci-dessous, même des appariements complémentaires bizarres apparaissent lors de la prédiction de la structure de l'ARN, tels que G:G, G:A, etc.
Limitations de l'utilisation d'AlphaFold 3
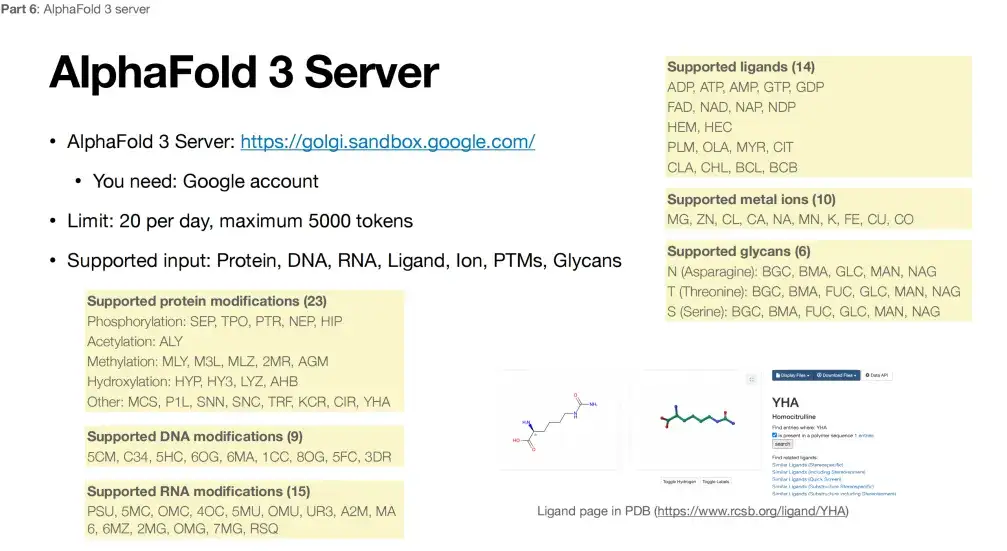
Partant du principe que les données ne sont pas hautement confidentielles, tout le monde peut accéder à AlphaFold 3 via le site Web fourni par Google. Cependant, la plateforme présente également certaines limites. Comme le montre la figure ci-dessous, en termes de modification des protéines, AlphaFold 3 ne prend actuellement en charge qu'un nombre limité de modifications à 3 positions spécifiques, totalisant 23 types. La modification de l'ADN ne prend en charge que 9 types, la modification de l'ARN ne prend en charge que 15 types, les ions métalliques ne prennent en charge que 10 métaux différents et les ligands sont limités à 14 petites molécules.
Par conséquent, compte tenu des limitations spécifiques mentionnées ci-dessus, AlphaFold 3 pourrait ne pas être en mesure de gérer la plupart des recherches et des réactions, et devra peut-être attendre qu'il soit véritablement open source.
En résumé, AlphaFold 3 a réalisé des progrès significatifs dans l’élargissement de sa portée de prédiction, surpassant les modèles d’IA existants, mais ses performances sur des tâches spécifiques doivent encore être améliorées, en particulier dans la prédiction de structures fines.Par conséquent, bien qu’AlphaFold 3 ait réalisé des progrès significatifs, des recherches et des efforts continus sont encore nécessaires pour résoudre complètement certains problèmes complexes.
À propos de Zhong Bo Zitao

Zhong Bozitao est actuellement doctorant en intelligence artificielle à l'Université Jiao Tong de Shanghai. Ses principales orientations de recherche comprennent la prédiction de la structure et de la fonction des protéines à haut débit, la génération de conformation des protéines, etc. Il a publié plus de 20 articles depuis 2019 et a publié les résultats de l'analyse de prédiction de la structure AlphaFold à haut débit de la relation entre le protéome des grands fonds et les voies métaboliques dans Nature Communications. Il a remporté la médaille d'or du Concours international de machines génétiquement modifiées (iGEM) à trois reprises et a été juge à plusieurs reprises.
Google Scholar:
https://scholar.google.com/cita








