Command Palette
Search for a command to run...
L'IA Pour La Génomique | Algorithme De Représentation Spatiale Des Données Du Transcriptome SPACE, Application De l'intelligence Artificielle En Génomique

Dans le deuxième épisode de la série « Meet AI4S », nous avons l'honneur d'inviter Li Yuzhe, chercheur postdoctoral au laboratoire de Zhang Qiangfeng à l'École des sciences de la vie de l'Université Tsinghua,Son laboratoire, Zhang Qiangfeng, appartient à l'École des sciences de la vie de l'Université Tsinghua. Il s'agit également d'une partie importante du Centre conjoint des sciences de la vie de l'Université Tsinghua-Pékin et du Centre d'innovation avancée de Pékin pour la biologie structurelle. Les recherches du laboratoire se concentrent sur l'intersection des sciences de la vie et des algorithmes d'intelligence artificielle, la technologie des groupes structuraux d'ARN et le développement d'algorithmes, la technologie et le développement d'algorithmes de séquençage du génome unicellulaire, la modélisation de la structure des protéines basée sur des données de cryomicroscopie électronique et le développement d'algorithmes d'intelligence artificielle associés.
Ce partage,Le Dr Li Yuzhe a prononcé un discours intitulé « Explorer les applications de l'IA en génomique : prendre l'algorithme de caractérisation des données du transcriptome spatial SPACE comme exemple ».Les derniers résultats de recherche de l’équipe ont été partagés et les méthodes d’IA dans la recherche en transcriptomique spatiale et en omique unicellulaire ont été présentées.
HyperAI a compilé et résumé le partage approfondi du Dr Li Yuzhe sans violer l'intention initiale.
Cliquez pour voir la rediffusion complète en direct :
L'IA pour la science apporte d'énormes changements au paradigme de la recherche dans le domaine scientifique
Aujourd’hui, le sujet que je vais partager est l’IA pour la science. Je crois que l’IA pour la science a apporté des changements majeurs au paradigme de la recherche dans l’ensemble du domaine scientifique. Ensuite, je l’expliquerai en détail en utilisant la recherche sur la structure des protéines comme exemple.
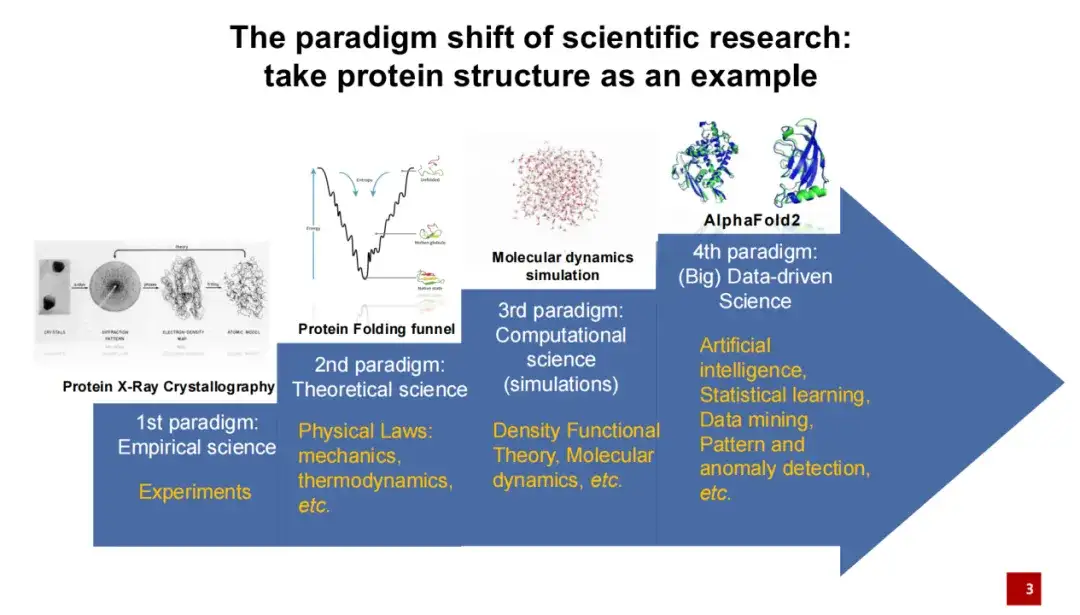
La première génération de paradigme de recherche sur la structure des protéines a été principalement menée par des moyens expérimentaux.C'est-à-dire, utiliser des rayons X pour photographier les cristaux formés par la protéine, puis effectuer une modélisation structurelle.
La deuxième génération de paradigme de recherche sur la structure des protéines a été principalement menée par des physiciens qui ont ajouté des connaissances théoriques à l’étude de la structure des protéines.Par exemple, si l’énergie de repliement d’une protéine est faible, alors ce repliement est relativement stable.
La troisième génération de paradigme de recherche sur la structure des protéines fait référence aux années 1990, lorsque, avec le développement de la technologie informatique, la simulation par ordinateur a été progressivement appliquée à la recherche sur la structure des protéines.En particulier, les simulations de dynamique moléculaire ont été largement utilisées ces dernières années. Ces méthodes de simulation nous aident à mieux calculer et prédire les structures des protéines dans une certaine mesure. Ces dernières années, notamment en 2020, les algorithmes d’intelligence artificielle sont entrés dans le domaine de la structure des protéines, apportant une nouvelle avancée. Lors du concours de prédiction de la structure des protéines 2020, AlphaFold 2 était loin devant les autres méthodes concurrentes.
L’introduction de l’intelligence artificielle a entraîné un changement de paradigme majeur dans les sciences de la vie et dans l’ensemble du domaine de la recherche scientifique. Par rapport aux méthodes de recherche traditionnelles,L’intelligence artificielle met davantage l’accent sur le fait de partir des données et de mener des recherches scientifiques basées sur ces données.Cela signifie que nous n’avons plus besoin de proposer une hypothèse scientifique à l’avance, mais que nous pouvons apprendre et révéler les lois de la nature directement à partir des données.
L'évolution de l'IA pour la génomique
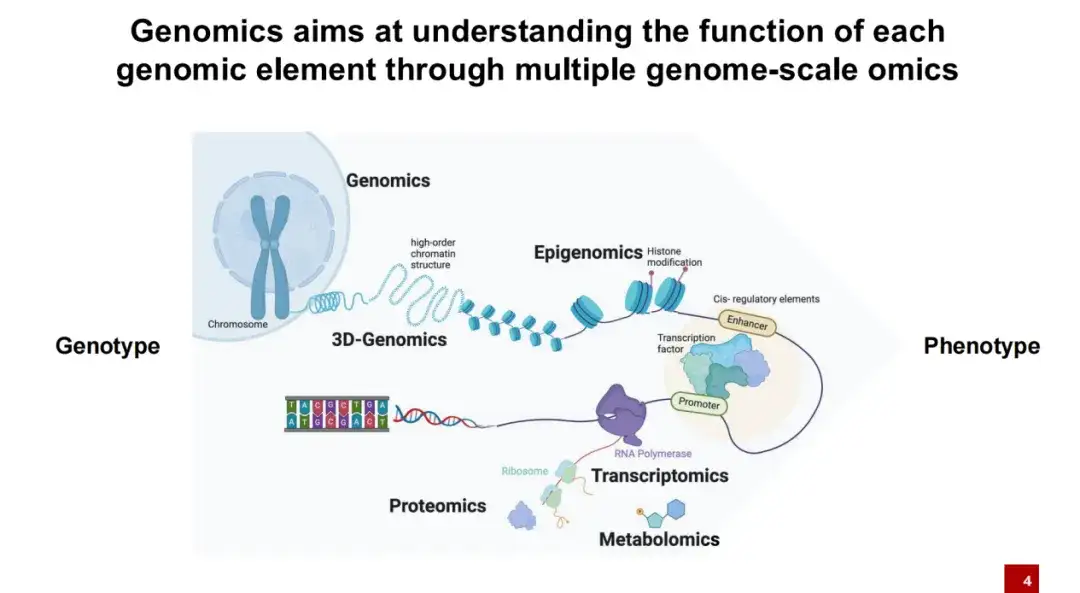
Le partage suivant portera sur l’application de l’IA dans le domaine de la génomique. en bref,La recherche en génomique explore principalement la relation entre le génotype (tout l’ADN du corps) et le phénotype (caractéristiques individuelles telles que la taille et le poids).
Comme nous le savons tous, l’ADN n’existe pas nu dans les cellules, mais est enroulé autour des nucléosomes. Les nucléosomes sont attachés à de nombreuses modifications d'histones. Normalement, ces ADN sont étroitement enroulés ensemble. Ce n'est que dans certaines conditions que l'ADN sera exposé pour former un intervalle ouvert. À ce stade, des protéines telles que les facteurs de transcription peuvent se lier à ces régions d’ADN exposées.
Dans le processus de transcription ultérieur, l'ARN peut être transcrit par l'ARN polymérase, puis traduit en protéine par le ribosome, et la protéine joue finalement un rôle dans les activités vitales.L’objectif de la recherche en génomique est de comprendre comment divers éléments de l’ADN affectent les activités de la vie.
Nous résumons les événements et développements importants dans le développement de l’IA pour la science depuis que la structure de la double hélice de l’ADN a été déchiffrée dans les années 1950 jusqu’à la période récente. Ses origines remontent à la découverte de la structure en double hélice de l’ADN dans les années 1950 et au développement de la technologie de séquençage Sanger dans les années 1970.
Comme le montre la figure ci-dessous, la partie bleue représente le développement de diverses technologies de séquençage et technologies expérimentales ; la partie verte représente les méthodes importantes dans le domaine de l'intelligence artificielle ; la partie jaune représente la mise en place de certains plans de recherche et bases de données importants à grande échelle ; la partie rouge représente les méthodes et applications représentatives dans le domaine de l'IA pour la génomique.
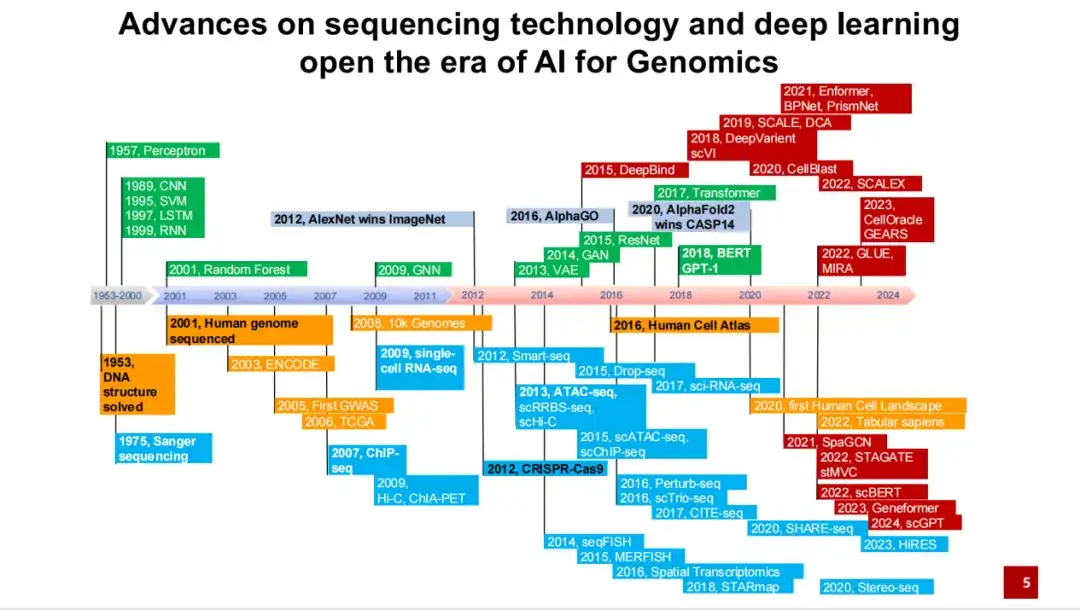
Comme vous pouvez le voir,2001, l'ébauche du projet génome humain a été initialement achevée, séquençant la séquence entière d'ADN d'un homme blanc. En 2012,AlexNet a surpassé les humains dans les tâches de classification d'images pour la première fois, inaugurant le développement explosif de l'intelligence artificielle au cours de la dernière décennie. En 2016,Le projet d’Atlas des cellules humaines a été proposé et la recherche est progressivement passée de la séquence d’ADN d’un seul individu à celle de toutes les cellules. La même année, AlphaGo, basé sur des méthodes d'apprentissage par renforcement, a vaincu les humains au jeu de Go.
Pour l'IA pour la génomique ou l'IA pour la science,Une avancée importante a été qu'en 2020, AlphaFold 2 a pris une large première place au CASP 14.Cela a conduit à l’application d’un nombre croissant de méthodes d’intelligence artificielle au domaine de la génomique.
dans,La génomique unicellulaire est une avancée majeure dans le domaine de la génomique ces dernières années.La recherche génomique traditionnelle implique généralement un séquençage en masse. Supposons que chaque ligne de la figure ci-dessous représente un type de cellule et que les lignes de différentes couleurs représentent différents types de cellules. Dans le passé, les méthodes de séquençage impliquaient de mélanger et de séquencer l’ensemble du tissu, ce qui rendait difficile de déterminer de quelle cellule spécifique provenait chaque ADN ou ARN. L’émergence de la technologie unicellulaire nous permet non seulement d’obtenir tout l’ADN ou l’ARN d’un tissu, mais également d’identifier la source cellulaire spécifique de cet ADN ou de cet ARN. Étant donné que les différents types de cellules ont des expressions génétiques différentes et remplissent des fonctions différentes, nous pouvons mieux comprendre les activités de la vie.
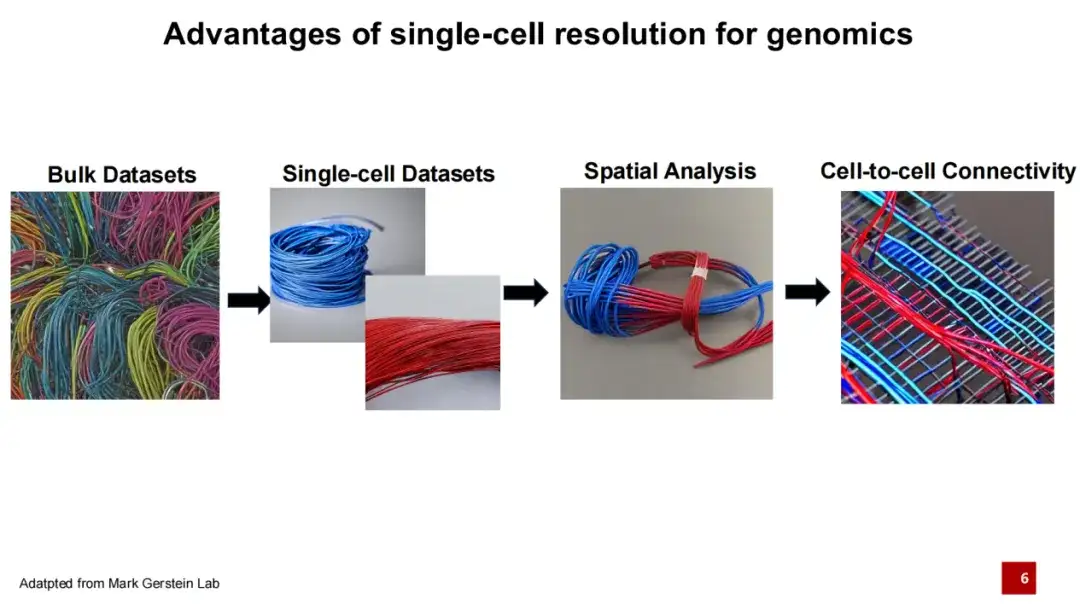
Au cours des cinq dernières années, la technologie omique spatiale représentée par la transcriptomique spatiale a franchi une étape supplémentaire en s'appuyant sur la technologie omique unicellulaire.Non seulement nous pouvons obtenir des informations sur chaque type de cellule, mais nous pouvons également déterminer la distribution de ces cellules dans l’espace.Étant donné que les interactions entre les cellules constituent une base importante pour la réalisation de leurs fonctions, des recherches plus poussées se concentrent sur la manière dont les cellules sont connectées.
Depuis le lancement du Projet Génome Humain jusqu'à la proposition du Projet Atlas des Cellules Humaines en 2016, son objectif a été de compléter la carte de référence de toutes les cellules humaines pour nous aider à mieux comprendre les activités de la vie et fournir un soutien au traitement et au diagnostic de maladies spécifiques.
L'équipe a développé trois méthodes : SCALE, SCALEX et SPACE pour la recherche en génomique unicellulaire
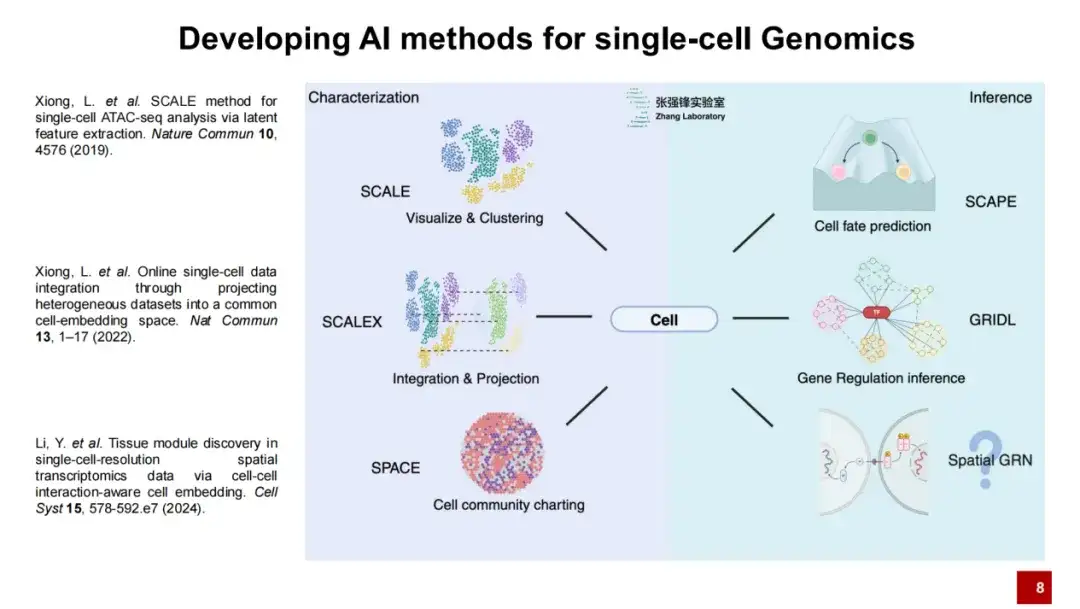
Notre laboratoire a développé une série de méthodes d’intelligence artificielle.Nous pensons que la génomique unicellulaire nécessite deux étapes principales : premièrement, la description de la cellule, et deuxièmement, l’inférence de la cellule.
Nous avons publié trois ouvrages pour décrire les cellules : SCALE, SCALEX et SPACE.SCALE est principalement destiné à la visualisation et au clustering, SCALEX à l'intégration et à la projection des données, et SPACE à la description de l'ensemble du microenvironnement d'organisation des données du transcriptome spatial.Aujourd'hui, je vais principalement présenter les deux méthodes : SCALEX et SPACE.
Méthode SCALEX pour éliminer les effets de lot
La méthode SCALEX consiste à éliminer les effets de lot.Il s’agit d’une question très importante dans la recherche en génomique. L'effet de lot fait référence aux différences dans les résultats expérimentaux entre différents lots en raison de facteurs techniques tels que des conditions expérimentales différentes.
Comme le montre la figure ci-dessous, même si nous cultivons deux répliques biologiques de cellules séparément, le séquençage de ces deux groupes de cellules devrait théoriquement produire une expression génétique très similaire. Cependant, pour des raisons techniques, telles que des différences d'environnement culturel, de temps de construction de la bibliothèque ou de plate-forme de séquençage,Les profils d’expression génétique finaux peuvent varier considérablement, introduisant ainsi beaucoup de bruit technique.Par conséquent, lors de l’analyse des données, il est nécessaire de supprimer cet effet de lot.

Dans la recherche biologique, les données ne peuvent souvent pas être collectées toutes en même temps, mais sont accumulées progressivement au travers de multiples expériences. donc,Supprimez les effets de lot et analysez les données de manière intégrée pour trouver les facteurs véritablement pertinents sur le plan biologique.Il s’agit d’une étape clé dans la recherche en génomique ou en génomique unicellulaire.
Sur cette base, nous avons développé la méthode SCALEX.Il est capable de projeter les données monocellulaires traitées dans un espace latent cellulaire généralisé. Le cadre de SCALEX est basé sur des autoencodeurs variationnels (VAE).
La première entrée est constituée des données du transcriptome d'une cellule unique, qui sont ensuite projetées dans un espace latent cellulaire généralisé via un encodeur sans lot.
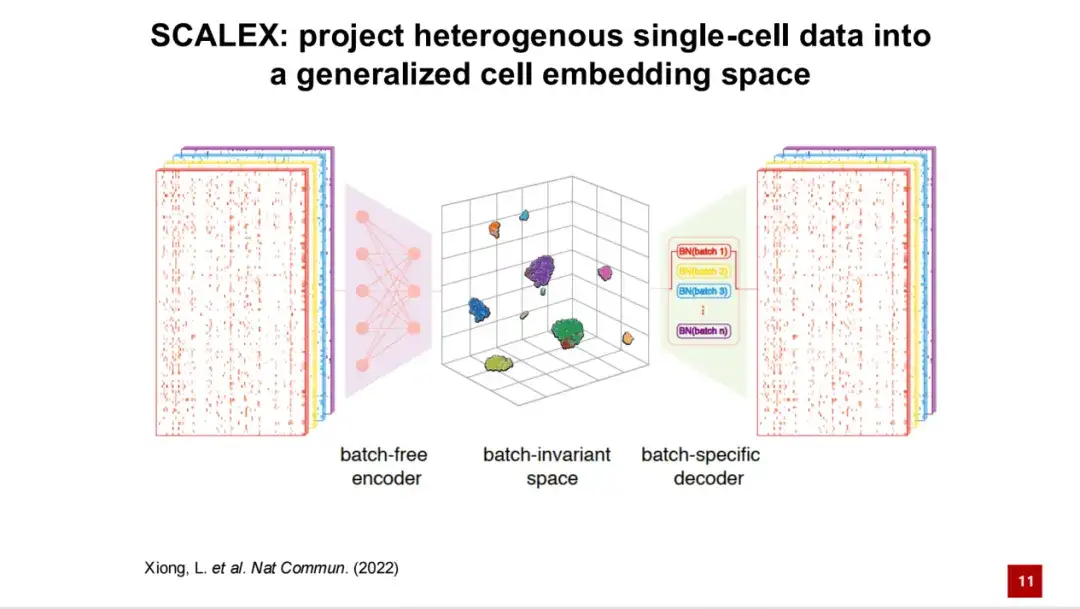
Ensuite, les informations de lot sont ajoutées au modèle via une normalisation de lot spécifique au domaine via un décodeur spécifique au lot. Grâce à cette conception asymétrique, l'espace latent cellulaire généré est un espace indépendant du lot, qui ne contient théoriquement aucun bruit technique lié au lot. L'expression génétique est reconstruite via le décodeur et la perte est calculée avec le spectre d'expression génétique d'entrée d'origine. En même temps, combinée à la divergence KL, la fonction de perte du modèle SCALEX est construite, qui est un modèle auto-supervisé.
Cette conception d'encodeur et de décodeur asymétrique présente deux avantages principaux :Premièrement, l’encodeur résultant est universel.Autrement dit, de nouvelles données peuvent être directement projetées dans l'espace latent de la cellule sans informations de lot via l'encodeur sans réentraîner le modèle ni réintégrer les nouvelles données dans les données existantes.
Deuxièmement, SCALEX accorde plus d’attention à l’effet de lot global.La méthode traditionnelle de suppression des effets de lot consiste principalement à rechercher des cellules similaires (paires de cellules) dans deux lots de données et à les associer pour correction afin de supprimer l'effet de lot. Cette méthode est essentiellement une correction d’effet de lot relativement locale.
Cependant, il existe un problème avec ce type de méthode : dans l’analyse réelle des données, les types de cellules de deux lots différents peuvent ne pas être complètement cohérents, il peut n’y avoir que quelques types de cellules en commun et le reste est spécifique au lot. Si l'appariement des cellules est forcé, une surcorrection peut se produire car les cellules appariées appropriées ne peuvent pas être trouvées et les types de cellules qui ne doivent pas être alignés seront forcés de s'aligner.
À cet égard, je vais expliquer plus en détail les deux principaux avantages de SCALEX.
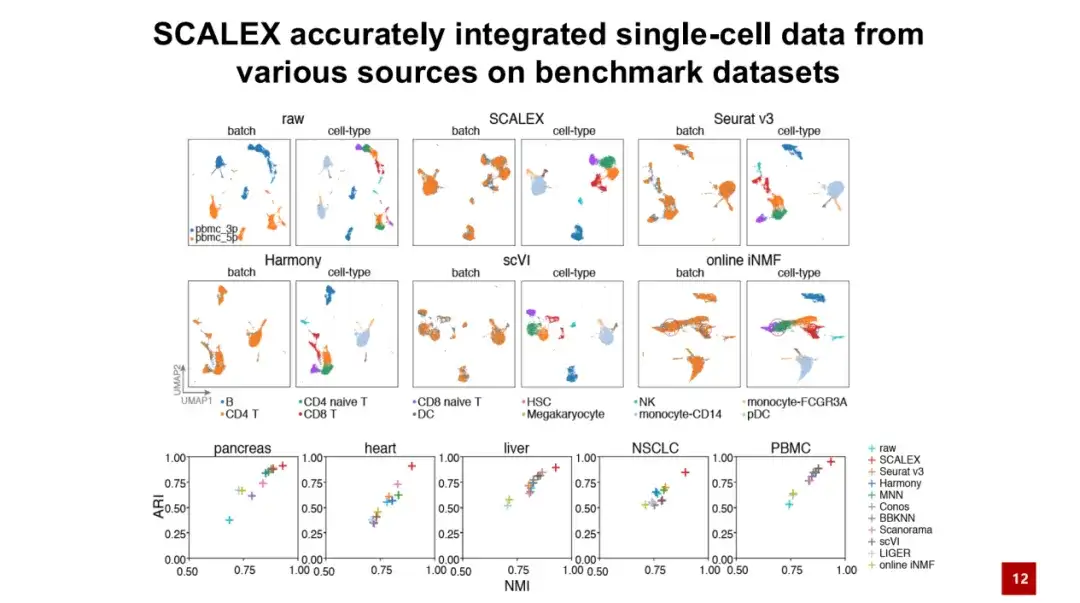
Tout d’abord, nous évaluons SCALEX sur cinq ensembles de données de test.Les résultats montrent que SCALEX surpasse les méthodes existantes en termes de précision.
Comme le montre la figure ci-dessous, le graphique par lots représente les données originales et non corrigées, le bleu et l'orange représentent respectivement 2 lots de données et le type de cellule représente le type de cellule. On peut constater que même s'il existe des types de cellules similaires dans ces deux lots, en raison de l'effet de lot important, les cellules qui appartenaient à l'origine au même type de cellule n'ont pas pu être agrégées ensemble, ce qui a entraîné des facteurs techniques masquant les facteurs biologiques et rendant impossible la recherche biologique ultérieure.
Après l’intégration de SCALEX, les deux lots de cellules ont pu bien s’agréger et ont été clairement séparés selon les types de cellules, démontrant l’importance de SCALEX dans les applications pratiques.
Un avantage important de SCALEX est qu’il peut traiter deux lots de données avec les mêmes types de cellules.Ces données sont appelées ensembles de données à chevauchement partiel. Comme le montre la figure ci-dessous, un chevauchement de 0 signifie que les types de cellules dans les deux lots sont complètement différents, et un chevauchement de 4 signifie qu'il existe 4 types de cellules partagés dans les deux lots.
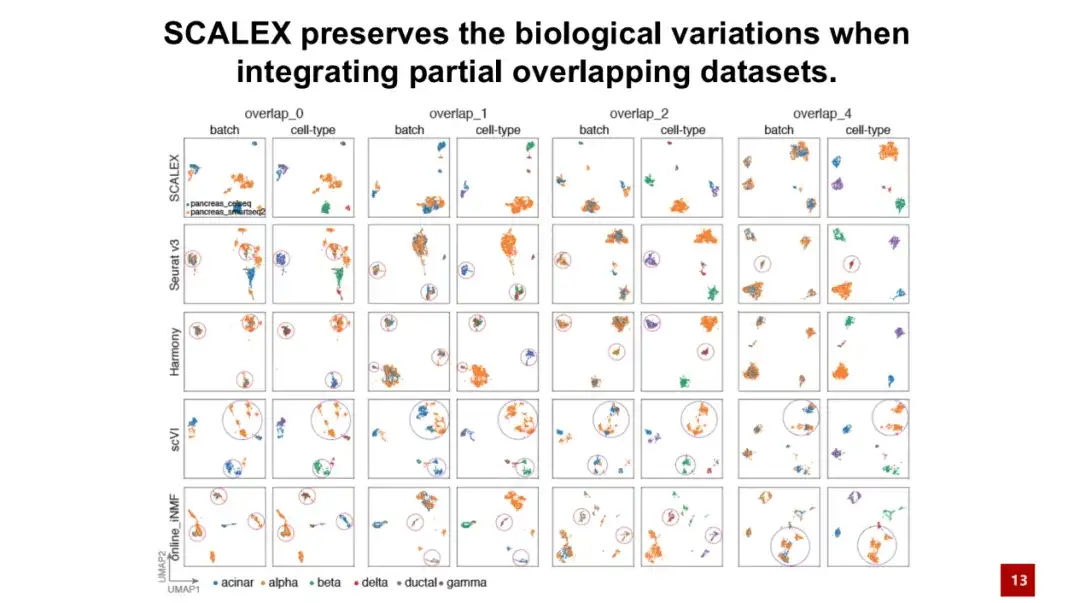
Les résultats montrent que même lorsque les deux lots de cellules n'ont aucun type cellulaire identique, SCALEX peut toujours bien maintenir les différences biologiques, c'est-à-dire que SCALEX n'intégrera pas de force des cellules de différents types cellulaires ensemble, tandis que d'autres méthodes similaires peuvent s'appuyer sur la recherche d'appariements de cellules, ce qui entraîne une surcorrection.
Un autre avantage de SCALEX est que l’encodeur universel peut projeter directement de nouvelles données dans l’espace latent existant des cellules sans effets de lot, sans réentraîner le modèle.Comme le montre la figure ci-dessous, un atlas cellulaire de référence est d’abord formé à l’aide des ensembles de données du pancréas, puis les trois nouvelles données sont directement projetées dans l’espace latent cellulaire via l’encodeur formé. Les couleurs de la figure représentent les types de cellules et les points gris représentent les types de cellules de référence construits. On peut constater que les différents types de cellules sont bien séparés dans leurs positions respectives sur la figure.
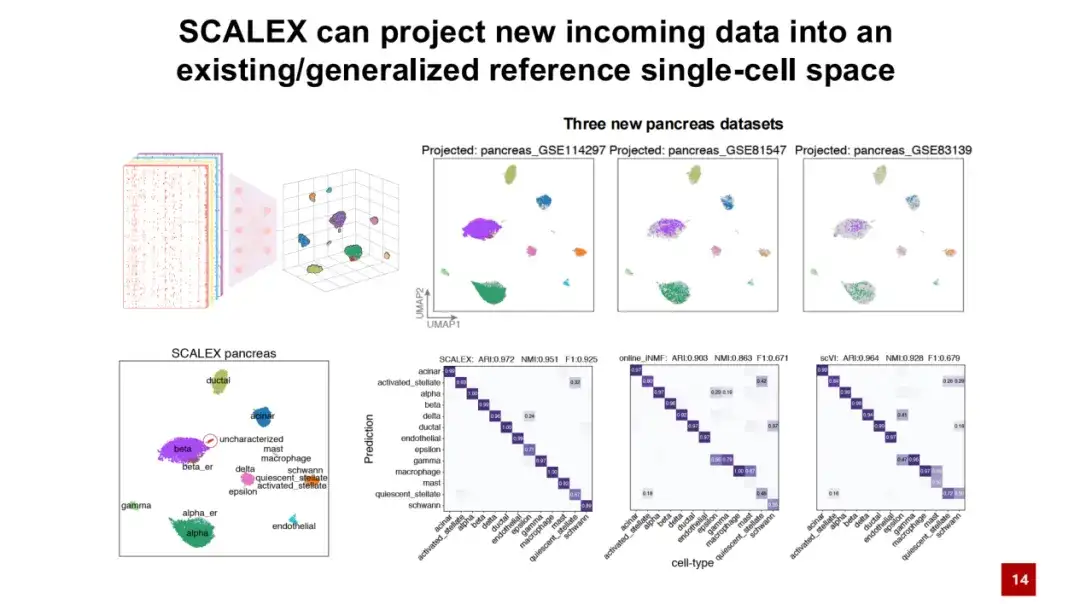
En projetant les étiquettes des cellules de référence autour d'un emplacement spécifique sur de nouvelles cellules de données, nous pouvons voir que SCALEX fonctionne bien dans l'annotation automatique des types de cellules. Comparé à d’autres méthodes existantes, SCALEX a une application très importante.Autrement dit, de nouvelles données peuvent être directement projetées dans les données construites, ce qui nous aide à effectuer une analyse comparative entre les données.
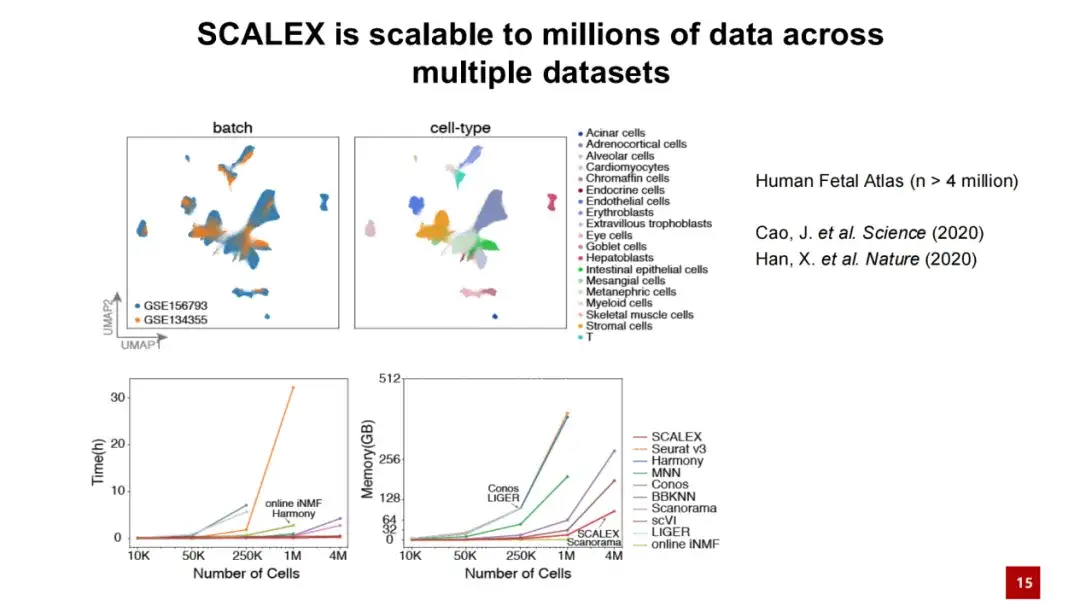
De plus, SCALEX est également performant dans le traitement de données à grande échelle. La figure ci-dessous montre que lorsque SCALEX traite 4 millions de données cellulaires, le temps de calcul ne dépasse pas quelques dizaines de minutes et la consommation de mémoire est inférieure à 100 Go. Cela montre que SCALEX présente une bonne évolutivité et peut être utilisé pour l’analyse intégrée de données monocellulaires à très grande échelle.
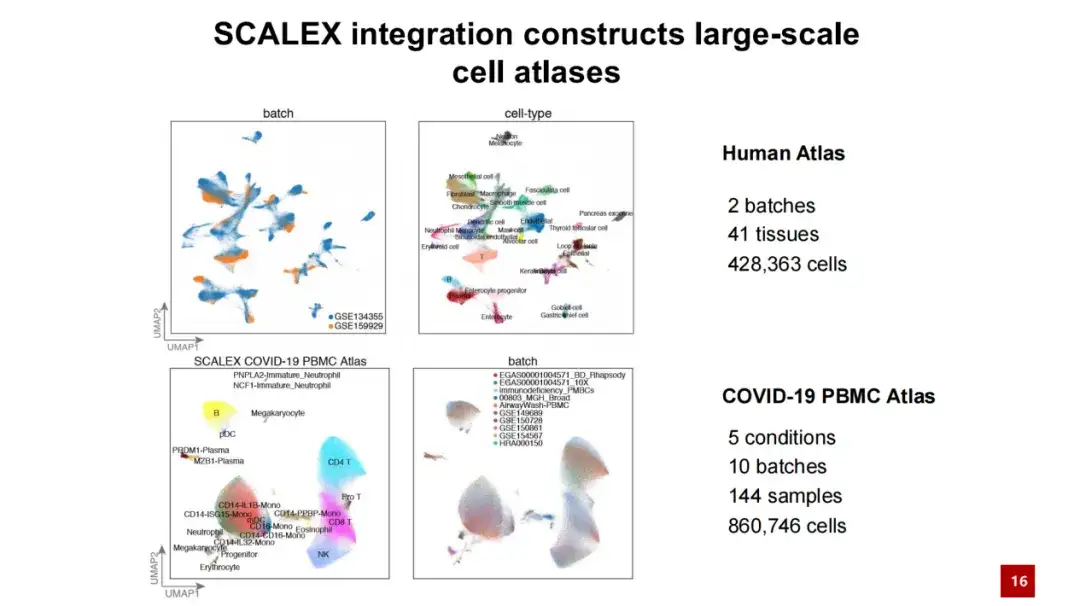
En profitant de SCALEX, nous avons construit deux atlas cellulaires à grande échelle, l’un est un atlas cellulaire d’individus humains, contenant plus de 400 000 cellules ; l'autre est un atlas cellulaire PBMC COVID-19, contenant plus de 860 000 cellules et plus de 100 échantillons.
SPACE : un outil d'analyse par intelligence artificielle des données de transcriptome spatial
Ensuite, je vous présenterai l’outil d’analyse du transcriptome spatial SPACE que mon équipe a récemment publié.
En termes simples, la technologie de transcriptomique spatiale peut fournir des informations sur l’expression génétique des cellules et leur emplacement spécifique dans l’espace. La figure ci-dessous montre un résultat typique de transcriptome spatial. Dans la figure de gauche, chaque point représente une cellule et la couleur indique son type de cellule. Ces cellules ont été regroupées par réduction de la dimensionnalité de l'expression génétique pour former une carte UMAP. Dans le panneau de droite, la position spatiale réelle de chaque cellule dans les données E16.5 de l'embryon de souris est affichée. On peut clairement voir que la distribution spatiale des cellules a une bonne spécificité.
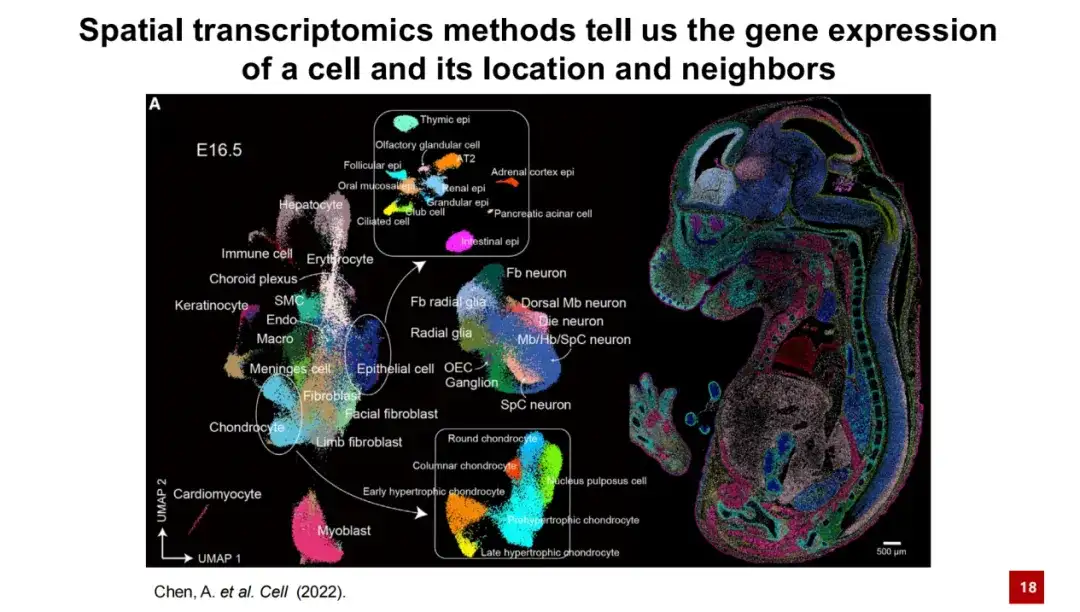
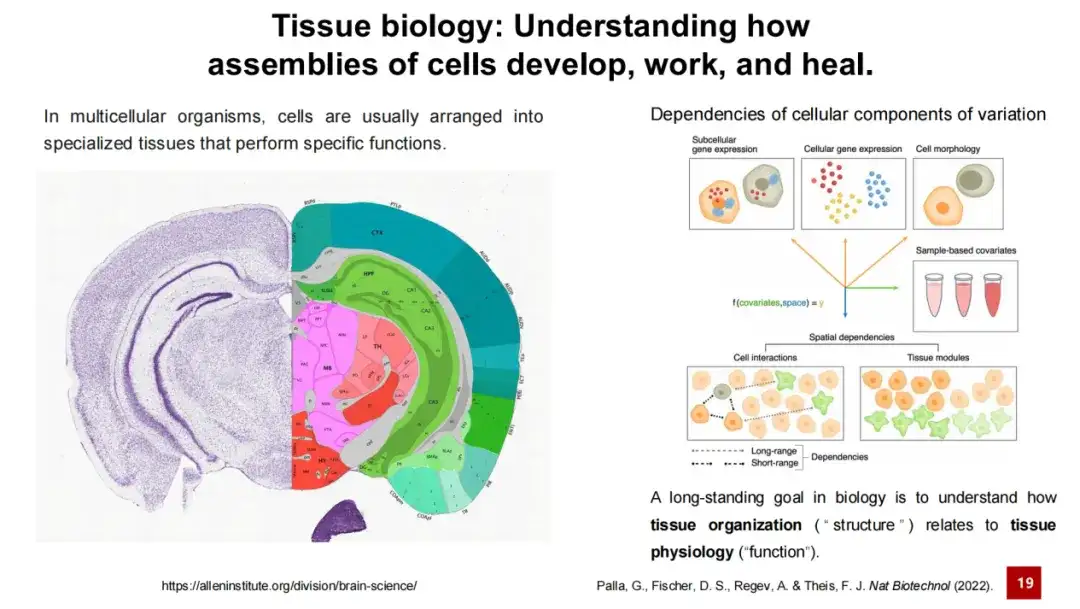
La recherche organisationnelle a toujours été l’un des enjeux fondamentaux de la recherche en sciences de la vie. On peut dire que l’un des objectifs à long terme de la recherche biologique est de comprendre la relation entre la structure d’une organisation et sa fonction. C'est facile à comprendre. Par exemple, différentes régions du cerveau sont composées de différents neurones et cellules de soutien, qui remplissent différentes fonctions grâce à des interactions cellulaires complexes. Par exemple, certaines zones sont responsables de la mémoire, d’autres de l’apprentissage et d’autres encore des réponses motrices.
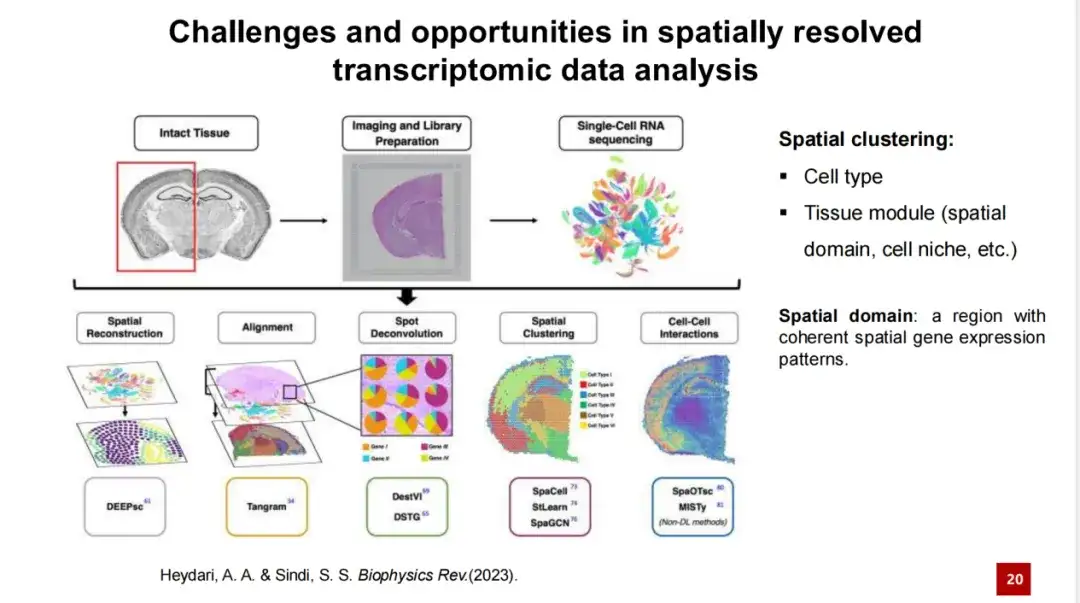
donc,L’un des problèmes fondamentaux de l’analyse du transcriptome spatial est d’identifier différents types de cellules ou modules tissulaires dans l’espace, une tâche collectivement appelée clustering spatial.
Cette tâche comprend deux sous-tâches : l’une consiste à identifier le type de cellule et l’autre à identifier le module tissulaire.. La première est plus intuitive, car elle permet d’identifier différents types de cellules dans les données du transcriptome spatial, comme le montrent les données sur les embryons de souris ; tandis que ce dernier est relativement abstrait, impliquant l'identification de zones dans le tissu qui sont plus petites que la structure du tissu, qui peuvent avoir des fonctions spécifiques ou être composées de cellules.
Dans différentes études, les chercheurs donnent des noms différents aux modules tissulaires, tels que domaine spatial ou niche cellulaire, parmi lesquels le domaine spatial est un terme plus couramment utilisé. Certains chercheurs pensent que l’identification des modules organisationnels consiste à identifier des régions présentant des caractéristiques d’expression génétique spatiale cohérentes.
Cependant, ce concept a des limites. Par exemple, la figure A ci-dessous montre qu’il existe une différence significative dans l’expression des gènes entre deux régions, mais dans les figures B et C, la distribution de l’expression des gènes entre les régions n’est pas complètement nette et peut être confondue. Les figures B et C montrent des situations que le concept de domaine spatial ne peut pas résoudre.

Pour résoudre ce problème, nous proposons la méthode SPACE.Le problème du domaine spatial est résolu en apprenant des intégrations cellulaires sensibles à l'interaction.
SPACE utilise un framework d'autoencodeur graphique pour apprendre les intégrations de cellules de faible dimension.
Tout d’abord, nous saisissons les données du transcriptome spatial et construisons un graphique voisin basé sur la position spatiale de chaque cellule, c’est-à-dire en connectant les cellules voisines les plus proches de chaque cellule pour former un graphique. Dans la figure ci-dessous, les nœuds représentent des cellules et les caractéristiques des nœuds sont les caractéristiques d’expression génétique des cellules. Nous entrons le graphique voisin et les profils d'expression génétique dans l'encodeur de SPACE, qui se compose d'un réseau GAT à trois couches.
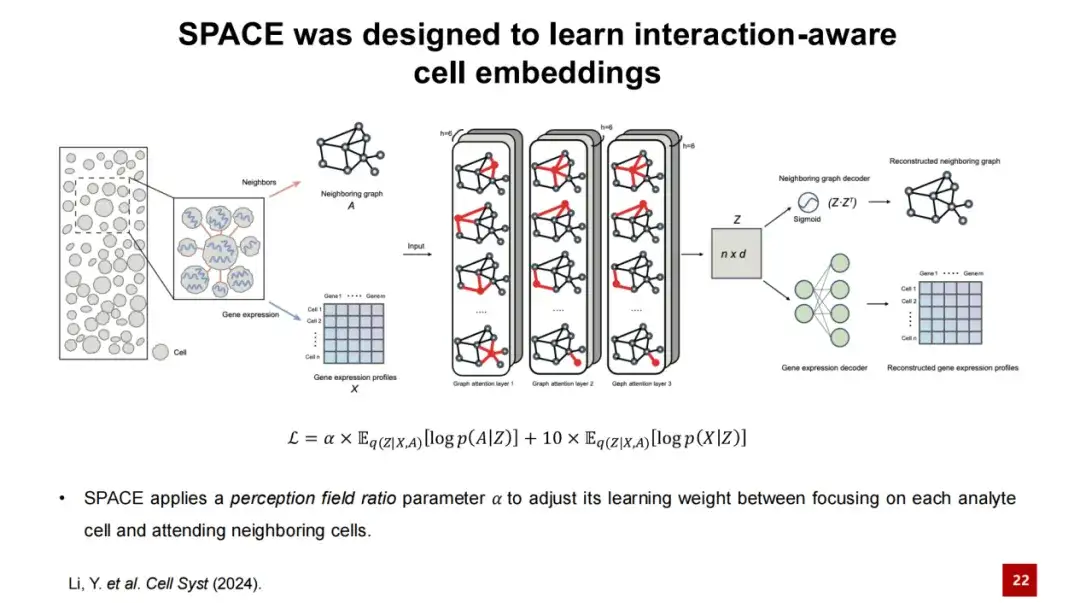
Grâce au traitement de l'encodeur, nous pouvons obtenir la représentation intégrée de chaque nœud et la reconstruire via deux décodeurs indépendants :Un décodeur reconstruit la représentation de la couche cachée de la cellule de faible dimension dans un graphique de voisinage, et l'autre décodeur reconstruit le profil d'expression génétique de la cellule. La fonction de perte du modèle SPACE est la somme de ces deux pertes de reconstruction.
Dans ce processus,Nous concevons un paramètre de rapport de champ de perception α pour ajuster les poids des deux fonctions de perte dans le modèle.
Lorsque la valeur α est petite,Le modèle se concentre davantage sur la reconstruction de l’expression génétique de la cellule elle-même, et l’intégration cellulaire obtenue peut être utilisée pour identifier le type de cellule ;Lorsque la valeur α est grande,Le modèle se concentre davantage sur l’interaction entre les cellules, et l’intégration cellulaire obtenue à ce stade peut être utilisée pour identifier les modules tissulaires. Étant donné que l'intégration cellulaire de faible dimension Z contient des informations sur les interactions cellulaires, nous appelons la représentation d'intégration cellulaire de faible dimension obtenue par interaction SPACE-awara.
Pour identifier les sous-types de cellules spatiales, nous avons appliqué SPACE à l’ensemble de données du cortex moteur primaire de la souris.
Dans la figure ci-dessous, le coin supérieur gauche montre la position spatiale de chaque cellule dans le tissu réel, où un point représente une cellule et la couleur indique le type de cellule. Il s’agit d’une carte UMAP générée en fonction de l’expression génétique. Les deux figures en bas à gauche montrent les sous-types de cellules spatiales identifiés par SPACE et leurs emplacements dans l’espace. Nous avons effectué une analyse de matrice de confusion de ces sous-types de cellules spatiales avec les types de cellules fournis dans l'étude originale (comme indiqué dans la figure de droite), et les résultats ont montré que les deux étaient généralement cohérents, avec un indice Rand ajusté (ARI) de 0,6. en même temps, SPACE peut différencier plus finement les astrocytes des oligodendrocytes et identifier davantage de sous-types cellulaires.
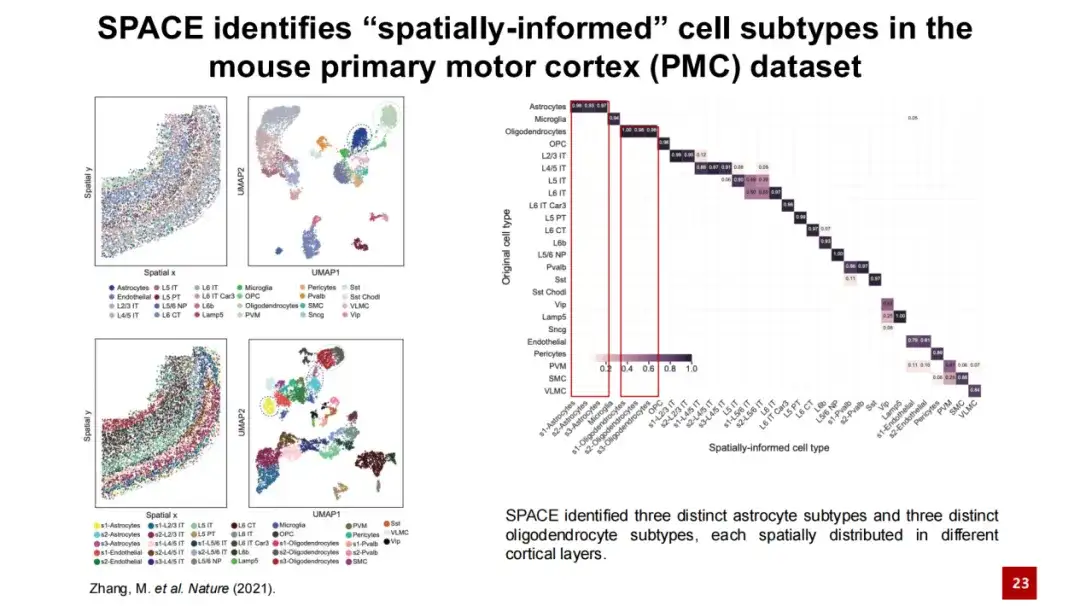
L'image en bas à gauche montre la structure organisationnelle du cortex moteur primaire des souris. La couche représente la structure corticale et la WM représente la matière blanche. La structure en couches de la couche 1 à la matière blanche est clairement visible. Les trois sous-types de cellules étoilées identifiés par SPACE étaient difficiles à distinguer par la seule expression génétique, et ils ont été mélangés dans la carte UMAP.
Cependant, ces trois sous-types cellulaires sont clairement distingués dans leur distribution spatiale : le sous-type cellulaire s1 est principalement distribué dans la zone allant de la couche 1 à la couche 4, s2 est principalement distribué dans la zone allant de la couche 5 à la couche 6 et s3 est principalement distribué dans la matière blanche. Nous avons compté les proportions de types de cellules entourant ces trois sous-types de cellules stellaires, et les résultats étaient cohérents avec cette règle de stratification. Bien que les trois sous-types cellulaires soient similaires en termes d’expression génétique, ils présentent néanmoins leurs propres gènes spécifiques à forte expression.

Les trois sous-types de cellules stellaires identifiés par SPACE sont très cohérents avec les études précédentes. Des études antérieures ont rapporté qu’il existe une interaction entre les astrocytes et les neurones, et que la stratification des astrocytes correspond à la stratification des neurones. En éliminant des facteurs clés dans les neurones, les chercheurs ont découvert que la structure en couches des neurones était détruite et que la structure en couches des astrocytes était également modifiée en conséquence. Cela suggère qu'il existe une interaction spatialement spécifique et une régulation génétique spatialement spécifique entre les astrocytes et les neurones.
À partir de cet exemple, nous pouvons voir queSPACE peut utiliser efficacement les informations spatiales et identifier avec précision différents types de cellules biologiques avec des caractéristiques spatiales.
L'article précédent a présenté que SPACE modifie la direction d'optimisation du modèle en ajustant le paramètre de rapport de champ de perception α : il peut prêter plus d'attention aux caractéristiques des cellules elles-mêmes pour identifier les types de cellules, ou prêter plus d'attention aux informations d'interaction entre les cellules pour découvrir les modules tissulaires.
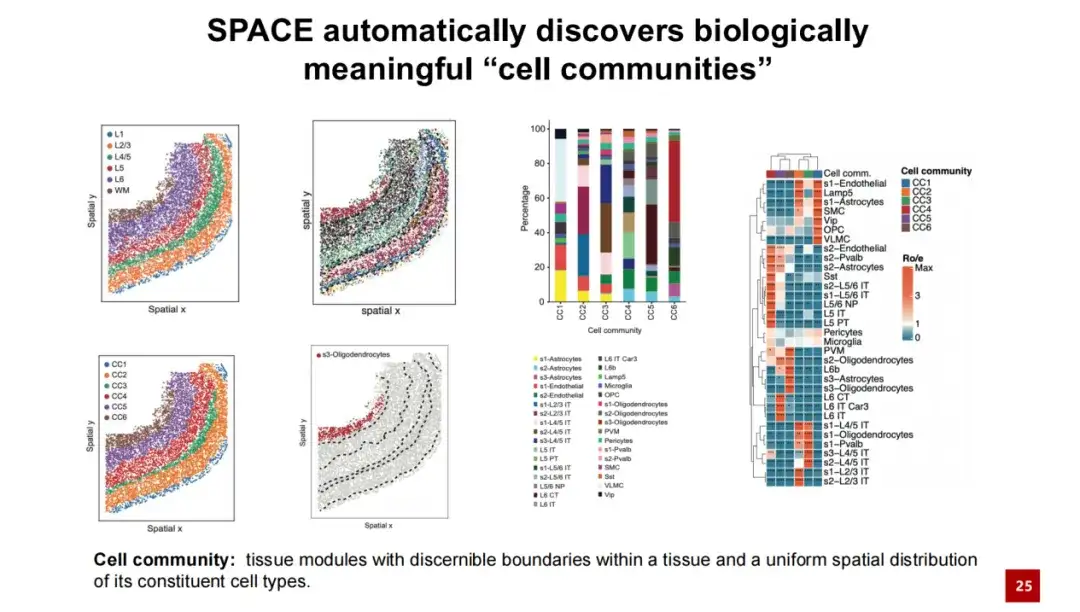
Dans le même ensemble de données,En augmentant la valeur α, SPACE a découvert avec succès le module tissulaire.Nous les avons appelés communautés cellulaires (CC en abrégé). Nous pensons que les modules tissulaires découverts par SPACE ont des limites identifiables et que la distribution spatiale des types de cellules en leur sein est relativement uniforme et cohérente. Nous avons comparé les communautés cellulaires découvertes par SPACE avec les structures tissulaires existantes et avons constaté que les deux présentent une bonne correspondance biunivoque. Chaque colonie cellulaire contient différents types de cellules et la distribution spatiale de ces types de cellules au sein de la colonie cellulaire est relativement uniforme.
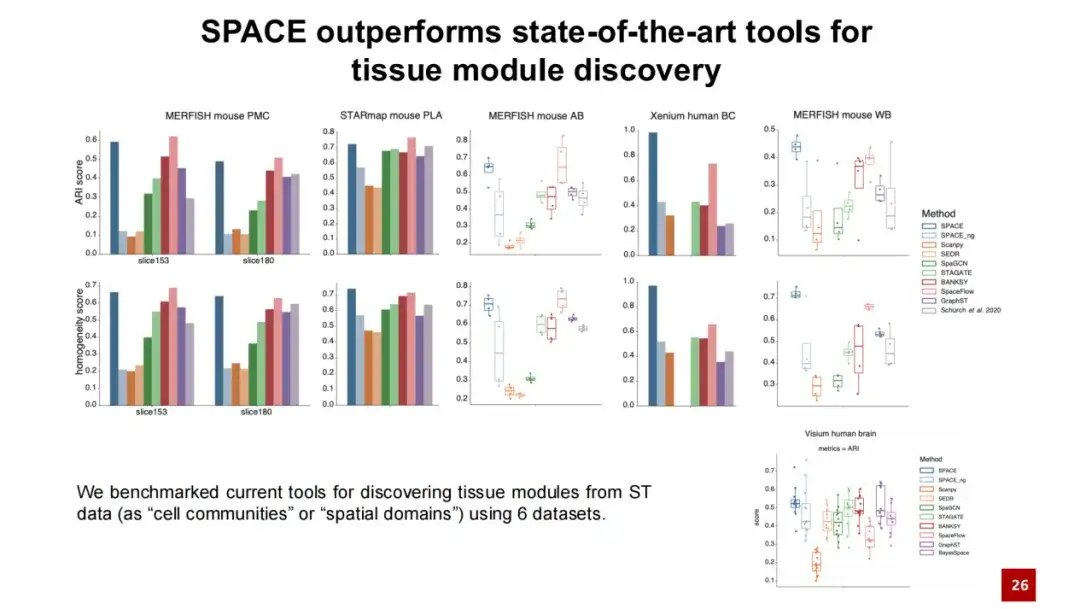
Nous avons comparé SPACE avec des méthodes existantes de découverte de modules organisationnels et effectué des tests sur cinq ensembles de données.Les résultats montrent que SPACE surpasse les meilleures méthodes existantes dans 2 ensembles de données et présente des performances comparables aux meilleures méthodes dans les 3 autres ensembles de données.Nous avons également effectué des tests et des analyses sur l’ensemble de données cérébrales humaines Visium couramment utilisé, et les résultats ont montré que SPACE est également applicable aux données de transcriptome spatial sans résolution unicellulaire.
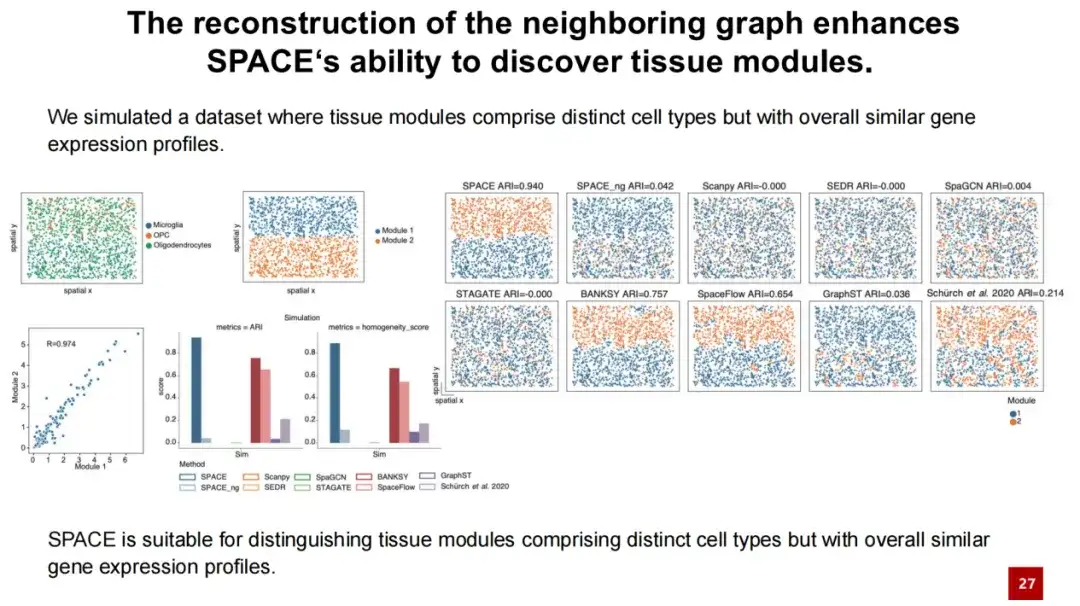
De plus, nous introduisons un modèle de test nommé SPACE_ng, où ng indique que nous désactivons la perte de reconstruction du graphe voisin dans le modèle SPACE. Les résultats montrent que les performances de SPACE_ng sont bien inférieures à celles de SPACE.
Pour illustrer davantage que SPACE peut bien découvrir les performances des modules organisationnels à partir de la reconstruction du graphe de voisinage, nous avons conçu une expérience de simulation. Nous avons sélectionné des oligodendrocytes et réparti uniformément la microglie et les OPC parmi les oligodendrocytes (voir l'image en haut à gauche ci-dessous), formant deux modules tissulaires.
Étant donné que la plupart des cellules de ces deux modules tissulaires sont des oligodendrocytes et présentent une très grande similarité (collaboration = 0,97), les résultats du test montrent que SPACE est bien supérieur aux autres méthodes, tandis que SPACE_ng ne peut pas faire la distinction entre les deux modules tissulaires.Cela indique que les performances du module de reconnaissance de tissus SPACE proviennent de sa reconstruction du graphe de voisinage.
Nous avons observé un phénomène similaire dans l’analyse en aval, c’est-à-dire que les caractéristiques des communautés cellulaires identifiées par SPACE ne se manifestent pas simplement par une expression génétique spatiale cohérente comme dans les domaines spatiaux, mais reflètent des interactions similaires entre les cellules voisines.
Dans la carte thermique ci-dessous, chaque colonne représente une cellule et sa couleur indique la population cellulaire à laquelle appartient la cellule et son type de cellule. Chaque ligne représente un type de cellule et montre la fréquence relative des interactions entre voisins entre ce type de cellule et d'autres cellules. À partir de cette carte thermique, nous pouvons voir que les cellules appartenant à la même communauté cellulaire présentent des similitudes dans les interactions entre voisins, et cette similitude est indépendante du type spécifique de cellules. En revanche, les cellules appartenant à différentes populations cellulaires ont montré de plus grandes différences dans leurs interactions avec leurs voisins.
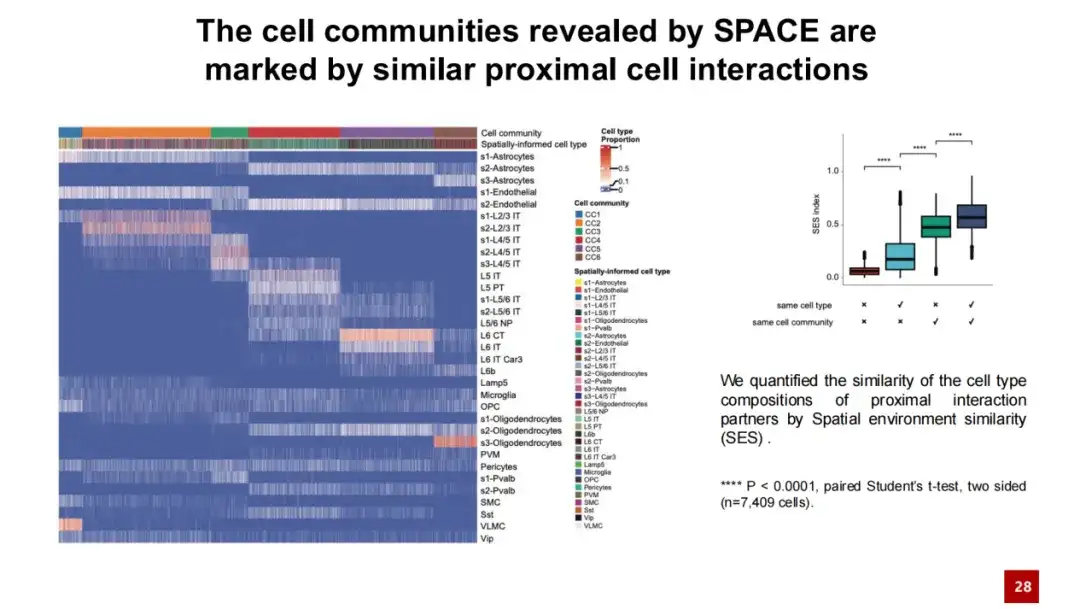
Nous avons également calculé quantitativement la similarité des interactions entre les cellules en utilisant la similarité cosinus. Les résultats ont montré que les cellules d’une même population cellulaire présentaient une grande similarité dans leurs interactions avec les cellules voisines, tandis que les cellules de différentes populations cellulaires présentaient des interactions extracellulaires relativement différentes. Ces résultats indiquent queLes communautés cellulaires découvertes par SPACE ne sont pas seulement un modèle d’expression génétique spatiale, mais sont également influencées par le réseau d’interaction cellulaire proximal.
Nous avons effectué une analyse similaire dans un autre ensemble de données de placenta de souris. L'image de gauche montre l'emplacement spatial de chaque type de cellule dans l'ensemble de données, l'image du milieu à gauche est la structure du tissu placentaire de souris annotée manuellement et l'image du milieu à droite montre les cinq populations cellulaires découvertes par SPACE. On peut constater qu’il existe une bonne correspondance biunivoque entre les communautés cellulaires découvertes par SPACE et les structures tissulaires annotées manuellement. Nous avons construit un réseau d'interaction cellulaire proximal caractéristique pour chaque population cellulaire, comme le montre la figure de droite, montrant les interactions cellule-cellule uniques au sein de chaque population cellulaire.

Prenons l’exemple de CC1, la communauté est principalement située dans la zone décidue maternelle. Nous avons constaté que dans CC1, il existe une forte interaction entre les cellules maternelles décapsidées S2 et les glycotrophoblastes S2. Des études antérieures ont montré que pendant la grossesse chez la souris, les glycotrophoblastes envahissent la région déciduale maternelle et interagissent avec les cellules déciduales maternelles qui s'y trouvent, déclenchant le remodelage des artères qui amènent le sang maternel au placenta, un processus essentiel à une grossesse normale.
De l’analyse ci-dessus, nous pouvons conclure queSPACE peut identifier les interactions cellulaires dans des échantillons biologiques qui ont des impacts importants sur les processus vitaux.Par conséquent, nous supposons queLes réseaux d'interaction construits par SPACE peuvent être utilisés pour optimiser l'analyse de la communication cellulaire basée sur le ligand-récepteur.
L'analyse de la communication cellulaire basée sur le ligand-récepteur est une méthode courante dans l'analyse des données unicellulaires, c'est-à-dire que, sur la base de l'expression génétique des ligands et des récepteurs de deux cellules, la possibilité de leur communication cellulaire via des paires ligand-récepteur est déduite. Nous avons d'abord analysé la communication cellulaire dans CC1 en utilisant CellChat, une méthode d'analyse de la communication cellulaire couramment utilisée, dans l'ensemble de données placentaires de souris.
CellChat a découvert que les cellules maternelles décapsidées s3 peuvent communiquer avec les types de cellules P-TGC via des voies de signalisation telles que le collagène FN1 et le THBS. Cependant, ces voies de signalisation nécessitent toutes un contact physique pour se produire réellement. Cependant, nous avons constaté que les deux types de cellules sont en réalité assez éloignés en termes de distribution spatiale (voir le coin inférieur droit de la figure ci-dessous), ce qui rend peu probable qu'ils entrent réellement en contact physique.
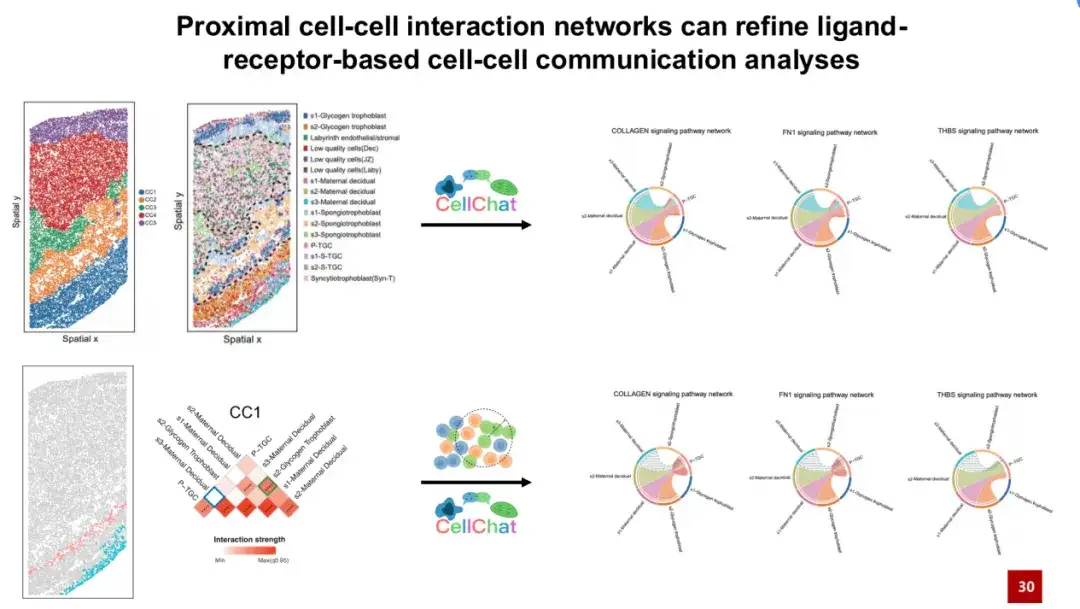
Cela a également été confirmé par le réseau d’interaction cellulaire proximal construit dans CC1. Les cases bleues montrent que les interactions entre elles sont peu probables.L'introduction du réseau d'interaction cellulaire proximal caractéristique construit par SPACE dans l'analyse de la communication cellulaire CellChat peut nous aider à exclure les signaux de communication cellulaire qui sont en fait impossibles à se produire dans l'espace, réduisant ainsi efficacement les faux signaux positifs.
Carrières
L'Université Tsinghua et le Laboratoire national clé de biologie membranaire ont créé une branche de biologie de la structure membranaire et de l'intelligence artificielle à Hangzhou. Actuellement, l’équipe recrute des professionnels engagés dans la recherche interdisciplinaire entre l’intelligence artificielle et la biologie. Nous invitons sincèrement les chercheurs intéressés par ce domaine à rejoindre l’équipe. Pour plus de détails sur le recrutement, veuillez scanner le code QR ci-dessous pour plus d'informations.









