Command Palette
Search for a command to run...
Sélectionné Pour La Conférence Principale De l'ACL2024 | InstructProtein : Aligner Le Langage Des Protéines Sur Le Langage Humain À l'aide d'instructions De Connaissances

En tant que base de la survie cellulaire, les protéines existent dans tous les organismes, y compris le corps humain. C'est l'échafaudage et la substance principale qui constitue les tissus et les organes, et joue un rôle essentiel dans les réactions chimiques essentielles à la vie.
Face à la complexité et à la variabilité de la structure des protéines, les méthodes expérimentales traditionnelles sont longues et laborieuses pour analyser la structure des protéines. Les modèles de langage des protéines (PLM) ont vu le jour. Ces modèles professionnels utilisent des séquences d’acides aminés comme entrée et peuvent prédire les fonctions des protéines et même concevoir des protéines complètement nouvelles. Cependant,Bien que les PLM soient très efficaces pour comprendre les séquences d’acides aminés, ils sont incapables de comprendre le langage humain.
De même, les grands modèles de langage (LLM) tels que ChatGPT et Claude-2, qui sont efficaces pour traiter le langage naturel, ne parviennent pas à décrire la fonction d’une séquence protéique ou à générer des protéines dotées de propriétés spécifiques. La raison est que,Les ensembles de données actuels sur les protéines et les textes présentent deux défauts majeurs : l’un est le manque de signaux d’instructions clairs ; l’autre est le déséquilibre de l’annotation des données. En résumé, il existe une lacune non comblée dans la recherche actuelle sur les LLM, à savoir l’incapacité à convertir rapidement le langage humain en langage protéique.
Pour résoudre ce genre de problème,L'équipe dirigée par Huajun Chen et Qiang Zhang de l'Université du Zhejiang a proposé le modèle InstructProtein, qui utilise des instructions de connaissances pour aligner le langage des protéines sur le langage humain.Nous avons exploré les capacités de génération bidirectionnelle entre le langage protéique et le langage humain, comblant efficacement le fossé entre les deux langages et démontrant la capacité d'intégrer des séquences biologiques dans de grands modèles linguistiques.
La recherche s'intitule « InstructProtein : aligner le langage humain et protéique via l'enseignement des connaissances ».Accepté par la conférence principale de l'ACL 2024.
Points saillants de la recherche :
* InstructProtein est une étude visant à aligner le langage humain et le langage des protéines grâce à des instructions de connaissance
* Exploré les capacités de génération bidirectionnelle entre le langage des protéines et le langage humain, comblant ainsi efficacement le fossé entre les deux langages
* Des expériences sur un grand nombre de tâches de génération de texte protéique bidirectionnelles montrent qu'InstructProtein surpasse les LLM de pointe existants

Adresse du document :
https://arxiv.org/abs/2310.03269
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : Ensemble de données scientifiques complet
Le corpus de la phase de pré-formation du modèle contient des séquences protéiques d'UniRef100 et des phrases provenant de résumés PubMed.Sur la base de ces données, les chercheurs ont généré un ensemble de données d’instructions contenant 2,8 millions de points de données.
Au cours de la phase de réglage fin du modèle, le graphe de connaissances sur les protéines a été construit à l'aide des annotations fournies par UniProt/Swiss-Prot, qui comprenaient les superfamilles de protéines, les familles, les domaines, les sites conservés, les sites actifs, les sites de liaison, les emplacements, les fonctions et les processus biologiques impliqués ; les données pour la modélisation causale des connaissances proviennent des bases de données InterPro et Gene Ontology.
Au cours de l'étape d'évaluation du modèle, les chercheurs ont sélectionné l'ensemble de données Gene Ontology (GO) pour évaluer la capacité du modèle à annoter la fonction des protéines, puis ont sélectionné Hu et al. ensemble de données pour évaluer la capacité du modèle à prédire la liaison des ions métalliques (MIB).
Architecture du modèle : Affiner le modèle pré-entraîné en créant un ensemble de données d'instructions de connaissances sur les protéines
Pour donner au LLM la capacité de comprendre le langage des protéines, InstructProtein adopte une approche de formation en deux étapes : d'abord une pré-formation sur des corpus de protéines et de langage naturel, puis un réglage fin avec un ensemble de données d'instructions de connaissances sur les protéines établi.
Phase de pré-formation
Au cours de la phase de pré-formation multilingue, cette étude a utilisé de grandes bases de données de textes liés à la biologie pour améliorer la compréhension linguistique du modèle et ses connaissances dans le domaine biologique. Le multilinguisme fait référence à la capacité de traiter à la fois les langues naturelles (comme les résumés en anglais) et les langages de séquences biologiques (comme les séquences de protéines).
Phase de réglage fin du modèle
Au cours de la phase de mise au point du modèle,Cette étude propose une méthode de construction d’ensembles de données appelée « instruction des connaissances ».Les graphiques de connaissances (KG) et les grands modèles de langage fonctionnent ensemble pour créer un ensemble de données d'instructions équilibré et diversifié. Cette méthode ne repose pas sur la capacité d’un grand modèle de langage à comprendre le langage des protéines, évitant ainsi les fausses informations introduites par un biais de modèle ou une hallucination. Le processus de construction spécifique est divisé en 3 étapes principales, comme le montre la figure suivante :
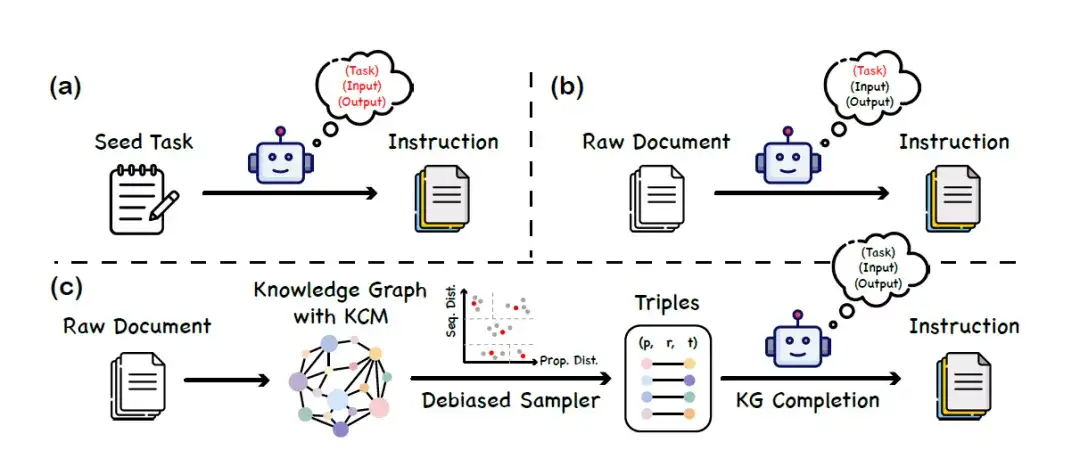
un. Étant donné un ensemble de tâches d'amorçage, demandez à LLM de générer de nouvelles données d'instruction
b. Utiliser LLM pour générer des données d'instructions correspondant au contenu du document brut
c. Cadre de génération d'instructions basé sur le graphe de connaissances (KG)
* Construction du graphe de connaissances :Les chercheurs ont utilisé UniProtKB comme source de données pour construire un graphique de connaissances sur les protéines. En empruntant le concept de pensée en chaîne, les chercheurs ont réalisé qu’il existe également des chaînes logiques dans l’annotation des protéines. Par exemple, les processus biologiques auxquels une protéine peut participer sont étroitement liés à sa fonction moléculaire et à sa localisation subcellulaire, et la fonction moléculaire elle-même est influencée par la structure du domaine protéique.
Pour représenter la chaîne causale de cette connaissance des protéines,Les chercheurs ont introduit un nouveau concept appelé Knowledge Causal Modeling (KCM).Plus précisément, le modèle causal de la connaissance se compose de multiples triplets interconnectés organisés dans un graphe acyclique dirigé, où la direction du bord représente la relation causale. Le graphique organise les triplets du niveau microscopique (couvrant les caractéristiques de la séquence protéique, telles que la structure) au niveau macroscopique (couvrant la fonction biologique). La figure ci-dessous montre le processus de génération d'instructions factuelles, logiques et diverses à partir d'un triplet contenant KCM en utilisant un grand modèle de langage combiné à un graphe de connaissances pour terminer la tâche.
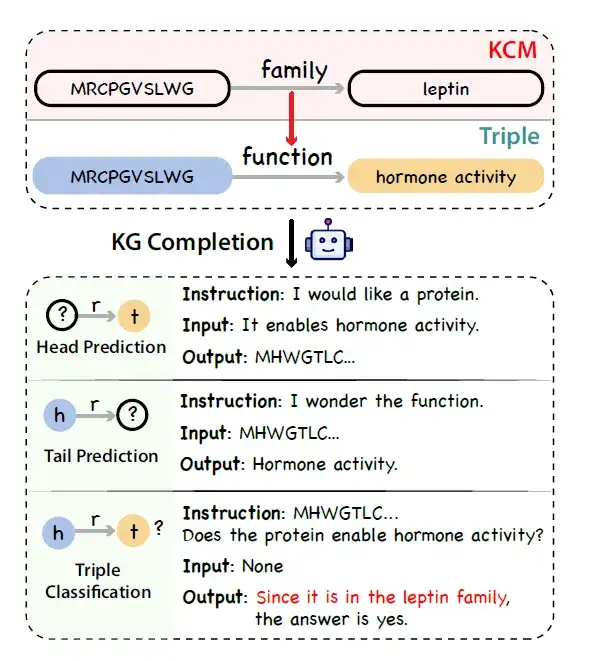
* Échantillonnage triple du graphique de connaissances :Compte tenu du problème de déséquilibre des annotations dans les graphes de connaissances, les chercheurs ont proposé une stratégie d’échantillonnage débiaisée pour remplacer l’échantillonnage uniforme comme stratégie alternative à l’échantillonnage uniforme. Plus précisément, il regroupe d’abord les protéines en fonction de leurs similitudes de séquence et d’attributs, puis extrait uniformément les triplets dans chaque groupe.
* Génération de données d'instructions :Les chercheurs ont simulé la tâche d'achèvement du graphique de connaissances et ont utilisé un LLM général (tel que ChatGPT) pour convertir les triplets du graphique de connaissances avec KCM en données d'instructions.
Cette approche permet la création efficace d’un ensemble de données riche et équilibré d’instructions sur la fonction et la localisation des protéines sans s’appuyer sur des modèles prédéfinis pour comprendre le langage des protéines.Fournir un support de données plus fiable pour la recherche et l’application ultérieures sur la fonction des protéines.
Grâce à une combinaison de pré-formation et de réglage fin, le modèle résultant, appelé InstructProtein, est capable de mieux effectuer diverses tâches de prédiction et d'annotation impliquant des séquences de protéines.Par exemple, prédire avec précision la fonction d’une protéine ou la localiser dans un emplacement subcellulaire spécifique – ce qui a des implications importantes pour l’ingénierie des protéines, la découverte de médicaments et la recherche biomédicale plus large.
Résultats de recherche : InstructProtein surpasse les LLM de pointe existants
L'étude a évalué de manière exhaustive les capacités d'InstructProtein en matière de compréhension et de conception de séquences protéiques :
Compréhension de la séquence des protéines
Les chercheurs ont évalué les performances du modèle InstructProtein sur les trois tâches de classification suivantes :Prédiction de l'emplacement des protéines, prédiction de la fonction des protéines, prédiction de la capacité de liaison des ions métalliques des protéines. Ces tâches sont conçues pour être similaires aux problèmes de compréhension de lecture en langage naturel, où chaque donnée contient une séquence protéique et une question, et le modèle doit répondre à une question de type oui/non. Toutes les évaluations sont effectuées dans un environnement sans tir.
Les résultats de l’évaluation sont présentés dans le tableau suivant :InstructProtein atteint de nouvelles performances de pointe sur toutes les tâches par rapport à tous les modèles de base.
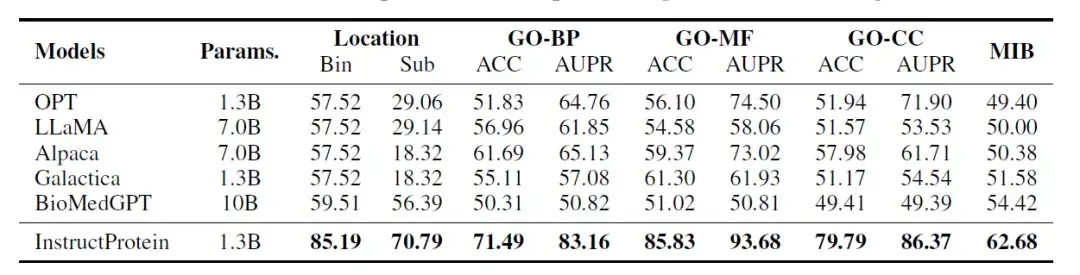
En outre, deux conclusions clés méritent d’être soulignées. Premièrement, InstructProtein surpasse considérablement les LLM dérivés de corpus de formation en langage naturel (c’est-à-dire OPT, LLaMA, Alpaca). Cela montre queLa formation avec un corpus contenant à la fois des protéines et du langage naturel est bénéfique pour les LLM et améliore leurs capacités de compréhension du langage des protéines.
Deuxièmement, bien que Galactica et BioMedGPT utilisent tous deux UniProtKB comme corpus pour le langage naturel et l’alignement des protéines, InstructProtein les surpasse systématiquement. Les résultats ont vérifié queLes données d’instruction de haute qualité de cette étude peuvent améliorer les performances dans les environnements à tir nul.
De plus, dans la tâche de localisation subcellulaire des protéines (bin), les LLM (OPT, LLaMA, Alpaca et Galactica) étaient gravement biaisés, ce qui a entraîné la classification de toutes les protéines dans le même groupe, ce qui a donné lieu à une précision de 57,52%.
Conception de séquences protéiques
En termes de conception de protéines, les chercheurs ont conçu une tâche de « couplage de protéines d'instruction » : étant donné une protéine et sa description, le modèle doit sélectionner la plus appropriée parmi sa description correspondante et 9 descriptions non correspondantes.
Comme le montre le tableau suivant :InstructProtein surpasse considérablement tous les modèles de base dans la tâche d'appariement instruction-protéine.
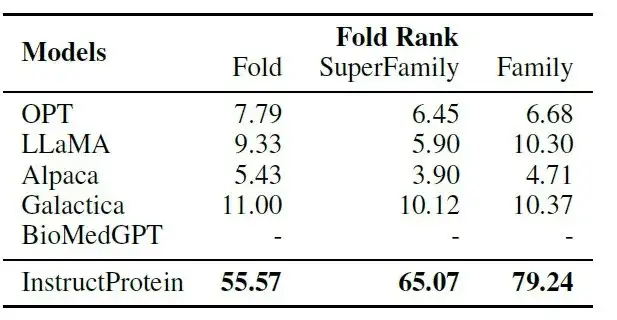
Parmi eux, BioMedGPT se concentre sur la conversion des protéines en texte et manque de capacités de conception de protéines ; Galactica a des performances limitées dans le cadre du zéro coup d'alignement des instructions avec les protéines car il est formé sur un corpus de protéines narratives.Ces résultats confirment la supériorité des capacités de suivi des instructions du modèle InstructProtein dans la production de protéines.
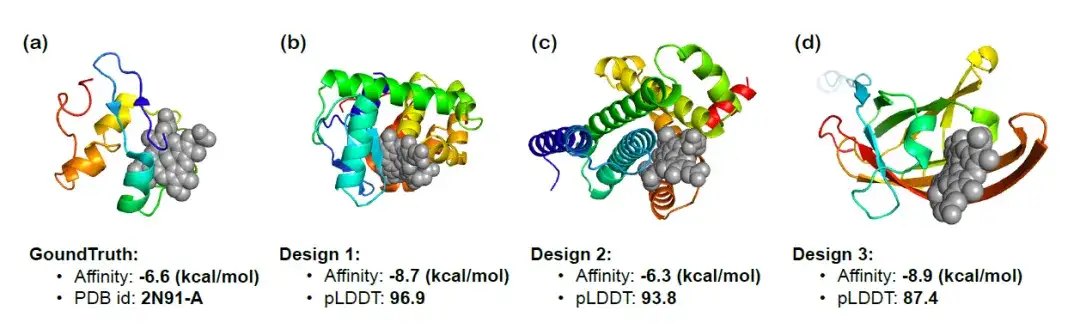
Pour valider davantage la capacité d'InstructProtein à concevoir des protéines suivant des instructions fonctionnellement pertinentes, les chercheurs ont utilisé InstructProtein pour concevoir des protéines de liaison à l'hème qui pourraient se lier à des composés spécifiques et visualisé les structures 3D des trois protéines résultantes. La figure ci-dessous montre les résultats de l'amarrage, la prédiction de l'affinité de liaison (plus elle est basse, mieux c'est) et le score pLDDT (plus la valeur absolue est élevée, mieux c'est). Il a été observé que la protéine résultante présentait une affinité de liaison significative,L’efficacité d’InstructProtein dans la conception de protéines de liaison à l’hème a été confirmée.
La route vers l’exploration des modèles protéiques ne fait que commencer
Ces dernières années, les grands modèles linguistiques ont apporté des changements révolutionnaires dans le domaine du traitement du langage naturel. Ces modèles sont largement utilisés dans de nombreux aspects de la vie quotidienne, tels que la traduction de langues, l’acquisition d’informations et la génération de code. Cependant, bien que ces modèles de langage soient performants dans le traitement du langage naturel et le codage du langage, ils sont incapables de gérer les séquences biologiques (telles que les séquences protéiques).Dans ce contexte, l’émergence d’un modèle de langage à grande échelle pour les protéines est opportune.
Le modèle de langage protéique est spécifiquement formé pour les données liées aux protéines, notamment les séquences d'acides aminés, les modèles de repliement des protéines et d'autres données biologiques liées aux protéines. Ils ont donc la capacité de prédire avec précision la structure, la fonction et les interactions des protéines. Le modèle de langage protéique représente l’application de pointe de la technologie de l’IA en biologie. En apprenant les modèles et les structures des séquences de protéines, il peut prédire la fonction et la morphologie des protéines, ce qui est d’une grande importance pour le développement de nouveaux médicaments, le traitement des maladies et la recherche biologique fondamentale.
En avril 2023, une étude publiée dans Science a montré que les chercheurs de l'équipe de méta-IA ont utilisé un grand modèle de langage capable de faire émerger des informations évolutives pour développer un prédicteur de séquence à structure ESMFold. La précision de prédiction pour les protéines à séquence unique dépassait celle d'AlphaFold2, et la précision de prédiction pour les protéines avec des séquences homologues était proche de celle d'AlphaFold2, et la vitesse était augmentée d'un ordre de grandeur. Le modèle a prédit plus de 600 millions de protéines métagénomes, démontrant l’étendue et la diversité des protéines naturelles.
En juillet 2023, Baidu Bio et l'Université Tsinghua ont proposé conjointement un modèle appelé xTrimo Protein General Language Model (xTrimoPGLM) avec un nombre de paramètres allant jusqu'à 100 milliards (100B). En termes de tâches de compréhension, xTrimoPGLM surpasse considérablement les autres modèles de base avancés dans une variété de tâches de compréhension des protéines ; en termes de tâches de génération, xTrimoPGLM est capable de générer de nouvelles séquences protéiques similaires aux structures protéiques naturelles.

Lien vers l'article :
https://www.biorxiv.org/content/10.1101/2023.07.05.547496v3
Juillet 2024Zhou Hao, chercheur associé à l'Institut des industries intelligentes de l'Université Tsinghua, a travaillé avec l'Université de Pékin, l'Université de Nanjing et l'équipe moléculaire de Shuimu pour proposer un modèle de langage protéique multi-échelle ESM-AA (ESM All Atom).En concevant des mécanismes de formation tels que l’expansion des résidus et le codage de position multi-échelle, la capacité de traiter des informations à l’échelle atomique a été élargie. Les performances de l'ESM-AA dans des tâches telles que la liaison cible-ligand ont été considérablement améliorées, surpassant les modèles actuels de langage protéique SOTA tels que l'ESM-2, et surpassant également les modèles actuels d'apprentissage de représentation moléculaire SOTA tels que Uni-Mol. Des recherches connexes ont été publiées lors de la principale conférence sur l'apprentissage automatique ICML sous le titre « ESM All-Atom : Multi-scale Protein Language Model for Unified Molecular Modeling ».

Adresse du document :
https://icml.cc/virtual/2024/poster/35119
Il convient de souligner que, bien que des progrès significatifs aient été réalisés dans la recherche sur les modèles de langage protéique à grande échelle, nous en sommes encore aux premiers stades de la compréhension complète de la complexité de l’espace de séquence des protéines. Par exemple, le modèle InstructProtein mentionné ci-dessus est confronté à des défis dans la gestion des tâches numériques, ce qui est particulièrement important dans le domaine de la modélisation des protéines qui nécessite une analyse quantitative, y compris l'établissement de la structure 3D, l'évaluation de la stabilité et l'évaluation fonctionnelle. avenir,Les recherches connexes seront étendues pour inclure une gamme plus large d’instructions, y compris des descriptions quantitatives,Améliorer la capacité du modèle à fournir des résultats quantitatifs, faisant ainsi progresser l’intégration du langage des protéines et du langage humain et élargissant sa praticité dans différents scénarios d’application.
Références :
1.https://arxiv.org/abs/2310.03269
2.https://mp.weixin.qq.com/s/UPsf9y9dcq_brLDYhIvz-w
3.https://hic.zju.edu.cn/ibct/2024/0228/c58187a2881806/page.htm








