Command Palette
Search for a command to run...
Dr. Bingxin Zhou De l'Université Jiao Tong De Shanghai : En Relevant Le Défi Des Données Biologiques Rares, Les Réseaux Neuronaux Graphiques Remodèlent La Compréhension Et La Génération Des Protéines

Le 12 août, l'école d'été sur l'IA pour la bio-ingénierie de l'Université Jiao Tong de Shanghai a officiellement ouvert ses portes, attirant plus de 100 initiés de l'industrie de plus de 30 universités et 27 entreprises nationales et étrangères. Au cours de ces trois jours d'études et d'échanges, de nombreux experts de l'industrie, représentants d'entreprises et jeunes chercheurs exceptionnelsUn partage approfondi a été proposé autour de l’intégration et du développement innovant de l’IA et de la bio-ingénierie.
Le 12 au matin, Zhou Bingxin, chercheur adjoint à l'Institut des sciences naturelles de l'Université Jiao Tong de Shanghai et au Centre national de mathématiques appliquées de Shanghai (branche de l'Université Jiao Tong de Shanghai), s'est exprimé sur le thème « Le passé et le présent de l'intelligence artificielle ». Il retrace de manière vivante l’histoire du développement de l’IA et résume les caractéristiques du modèle d’étape.
L'après-midi, dans le rapport d'expert invité de « Frontier Progress of Artificial Intelligence », le Dr Zhou Bingxin s'est également exprimé sur le thème « Réseaux neuronaux graphiques et représentation de la structure des protéines ». Nous avons partagé avec tout le monde la définition, les avantages et les applications de pointe des réseaux de neurones graphiques dans des domaines tels que la prédiction et la génération de protéines. HyperAI a compilé et résumé le discours du Dr Zhou Bingxin sur ce sujet sans violer l’intention initiale. Voici la transcription du discours.
Après des décennies de développement rapide, l'apprentissage profond a produit divers modèles tels que les réseaux neuronaux convolutifs, les réseaux neuronaux récurrents et Transformer, qui peuvent être utilisés pour traiter des données avec différentes caractéristiques. Parmi eux, les réseaux neuronaux graphiques sont largement utilisés dans divers scénarios tels que les réseaux sociaux, la prédiction de trajectoire, la modélisation moléculaire, etc. car ils peuvent saisir et traiter des données structurelles.
Cependant, de nombreuses personnes pensent que les réseaux neuronaux graphiques sont des réseaux convolutifs graphiques (GCN), qui ne peuvent pas s'adapter à des fonctions complexes et peuvent avoir des problèmes de lissage excessif lorsque plusieurs couches sont empilées, et présentent de nombreuses limitations. De plus, étant donné que les grands modèles basés sur Transformer ont de fortes capacités d’apprentissage sur de grands ensembles de données,Alors pourquoi continuons-nous à rechercher et à développer des réseaux neuronaux graphiques ?
À ces questions, je résume la réponse ainsi : « C'est SEXY ».
Le premier « S » est que la recherche basée sur les réseaux neuronaux graphiques est saine et durable. Comme le montre la figure ci-dessous, en comparant la consommation de carbone de divers comportements humains, on peut voir que la prémisse pour que le grand modèle ait des capacités puissantes est une énorme consommation d’énergie. De plus, la concentration excessive des ressources informatiques et la focalisation de la recherche sur de grands modèles réduiront également l’espace vital pour d’autres recherches sur les modèles. À long terme, seules les grandes entreprises qui monopolisent les ressources informatiques ou le pouvoir de parole seront en mesure de maintenir la recherche et le développement en intelligence artificielle, et l’espace de recherche scientifique pour les chercheurs des entreprises non grandes sera considérablement restreint.
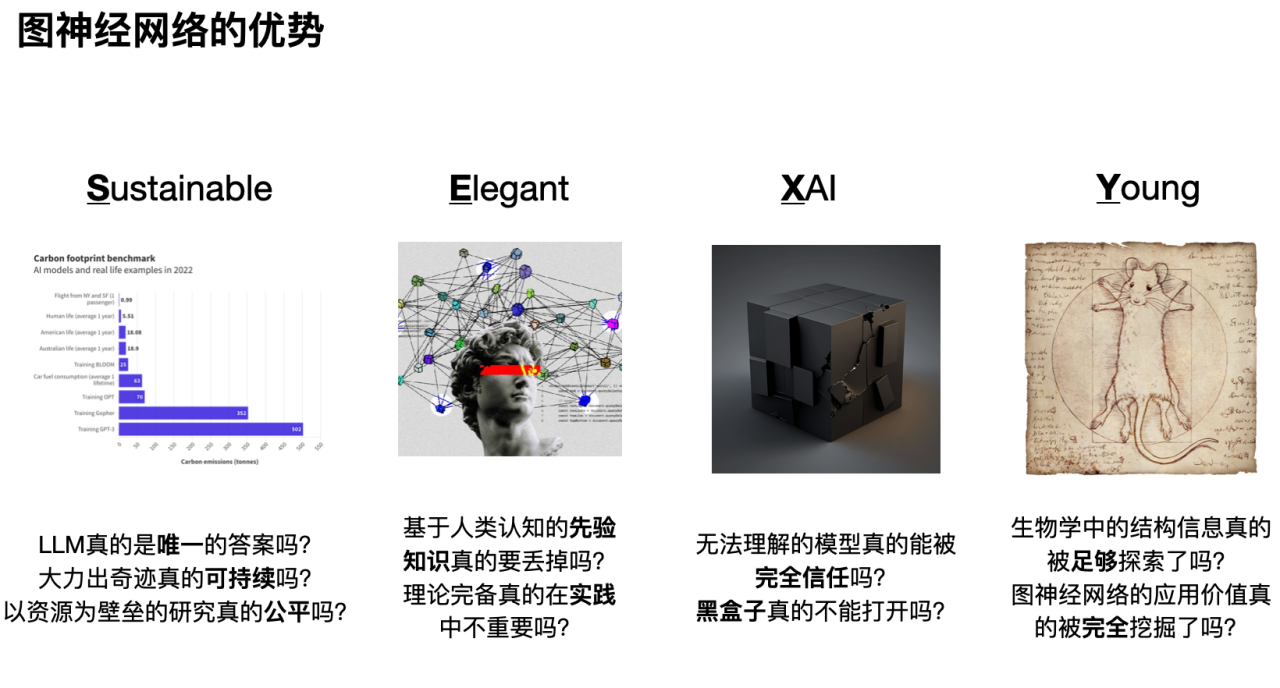
Le deuxième « E » est que des centaines d’années d’accumulation dans les sciences naturelles ne doivent pas être ignorées par le développement rapide de l’intelligence artificielle. En plus d'apprendre la représentation des caractéristiques, les réseaux neuronaux graphiques peuvent également intégrer de manière élégante les connaissances antérieures des humains (biais inductif). De plus, comparés à d’autres modèles basés sur les données, les réseaux neuronaux graphiques bénéficient d’un support théorique plus important, comme le traitement du signal, la dynamique sociale, etc.
Le troisième « X », les réseaux neuronaux graphiques, aident à promouvoir l’interprétabilité des réseaux d’apprentissage profond. Avec le développement de l’intelligence artificielle, les gens accordent de plus en plus d’attention à la signification et à la rationalité des résultats des modèles. Grâce à des recherches approfondies sur l’interprétabilité des réseaux neuronaux graphiques, nous pouvons mieux comprendre la logique et la base des décisions du modèle et améliorer la fiabilité et la confiance du modèle.
Quatrièmement, « Y » : En tant que domaine jeune et en développement rapide, les réseaux de neurones graphiques présentent encore un grand nombre de problèmes et de défis non résolus, offrant aux chercheurs un large espace d’exploration. De plus, tout comme les réseaux neuronaux convolutifs pour le traitement d’images et les mécanismes d’auto-attention pour le traitement du langage naturel, les réseaux neuronaux graphiques fourniront également de bonnes solutions à de nombreux problèmes biologiques (en particulier ceux avec des données insuffisantes et des connaissances préalables importantes).
Ensuite, je partagerai avec vous la valeur d'application spécifique des réseaux de neurones graphiques sous trois aspects : les données moléculaires et la représentation graphique, l'introduction aux réseaux de neurones graphiques classiques et les réseaux de neurones graphiques et d'autres problèmes biologiques.
Données moléculaires et représentation graphique : trois éléments des graphiques de données biologiques
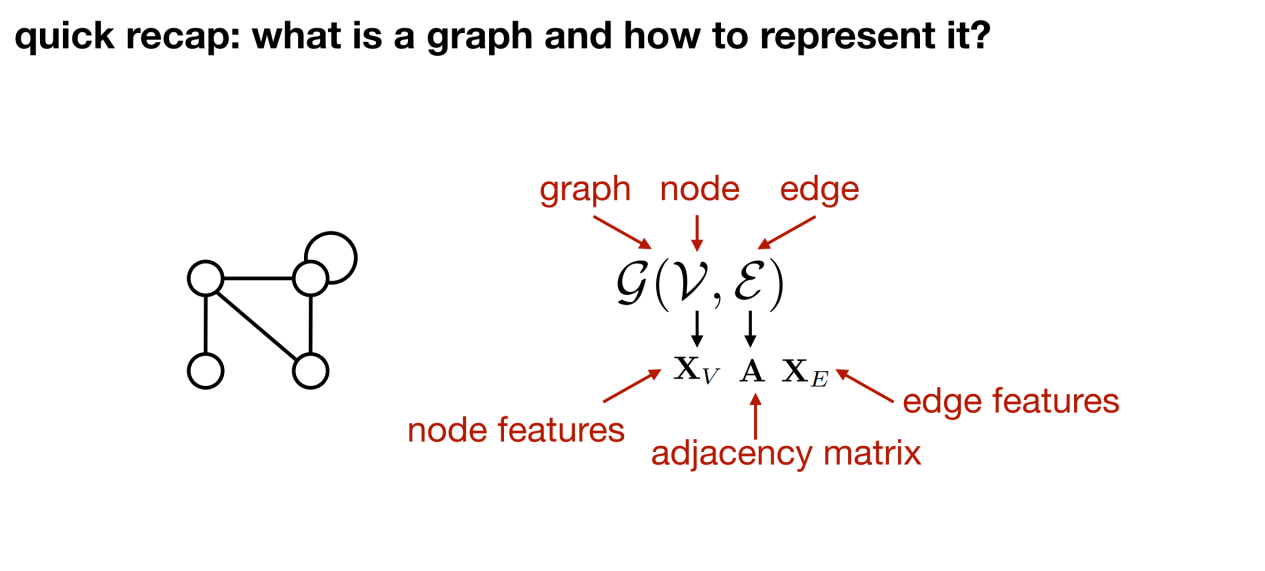
Si vous souhaitez convertir des données biologiques en une représentation graphique, vous devez d’abord répondre : qu’est-ce qu’un graphique et de quels éléments de base est-il composé ? En général,Un graphe contient trois éléments : les nœuds ; arêtes (relations de connexion entre les nœuds) ; graphes (l'entité complète composée de nœuds et d'arêtes).
Comment utilisons-nous ces trois éléments pour définir l’objet d’étude en biologie ? La figure suivante montre 4 cas :

Pour une petite molécule (Fig. Chaque atome peut être défini comme un nœud, et la relation de distance ou de liaison chimique entre les atomes peut être représentée par des arêtes.
Si nous examinons les protéines au niveau des acides aminés, La protéine dans son ensemble peut être visualisée sous forme de graphique, chaque acide aminé étant un nœud du graphique. Lorsque différents acides aminés sont proches en position spatiale, on peut supposer qu'il existe une certaine corrélation entre eux, et ces nœuds d'acides aminés spatialement proches sont reliés par des bords.
De même, si nous examinons les protéines en fonction de leur structure secondaire, Chaque structure secondaire peut alors être considérée comme un nœud dans le graphe protéique, et ses structures secondaires adjacentes ou spatialement proches sont reliées par des arêtes.
Enfin, pour le graphique de connaissances sur les maladies, Différentes maladies, gènes, médicaments, patients et autres éléments peuvent être considérés comme des nœuds, et les connexions entre les nœuds représentent les relations complexes entre eux, comme un certain médicament peut traiter une maladie spécifique, ou un certain gène provoque une certaine maladie.
Après avoir défini un graphique,L’étape suivante consiste à réfléchir à la manière de décrire les informations sur le graphique, telles que les caractéristiques des nœuds et des arêtes ?
Comme le montre la figure ci-dessous, il existe certaines relations entre les quatre nœuds. Afin de décrire avec précision ces relations, une matrice d'adjacence A peut être définie. Lors du traitement de différentes données biologiques, la matrice d'adjacence peut être utilisée pour caractériser s'il existe des liaisons covalentes entre les atomes ou pour déterminer les voisins d'ordre k d'un certain acide aminé.

De plus, chaque nœud et chaque arête peuvent être associés à un ensemble d’attributs. Prenons l’exemple d’un nœud d’acide aminé, les attributs du nœud peuvent inclure des informations caractéristiques telles que son type et ses propriétés physiques et chimiques. Les arêtes servent de ponts reliant les nœuds et peuvent également transporter des informations sur les fonctionnalités. Par exemple, le vecteur de caractéristiques sur chaque bord couvre la distance entre deux acides aminés (y compris la distance de séquence et la distance spatiale) et la base pour établir le bord (basée sur la structure spatiale ou les liaisons chimiques atomiques, etc.). Ces fonctionnalités de bord offrent une perspective plus détaillée et plus approfondie pour comprendre la relation entre les nœuds.
En résumé,Chaque entité structurée (comme une protéine) peut être représentée sous forme de graphique. Comme le montre la figure ci-dessous : G peut être utilisé pour représenter le graphique, v représente le nœud, ε représente l'arête, Xv représente les caractéristiques du nœud, la matrice d'adjacence A représente la connexion du nœud et Xe représente les caractéristiques de l'arête.
Sur la base des trois éléments de base d'un graphe (nœuds, arêtes et graphes), les tâches de représentation vectorielle et de prédiction sur le graphe peuvent être classées comme suit :
- Prédiction au niveau des nœuds. Par exemple, lors de la conception d'une séquence protéique, étant donné un graphique protéique connu, prédisez le type d'acide aminé représenté par chaque nœud du graphique.
- Prédiction de lien. Étant donné un graphique et tous les nœuds, déduisez s'il existe des relations entre les nœuds, telles que des réseaux de régulation génétique, des graphiques de connaissances sur les médicaments et d'autres tâches de prédiction.
- Prédiction graphique (prédiction au niveau du graphique). Lorsque les nœuds et les arêtes sont déterminés, plusieurs graphiques sont appris et analysés simultanément pour prédire les étiquettes de chaque graphique.
Que sont les réseaux neuronaux graphiques : pas seulement GCN, mais aussi GAT, GraphSAGE, EGNN, et bien d'autres
Les réseaux neuronaux graphiques recherchent des représentations de couches cachées de chaque nœud en fonction des relations de connexion entre les nœuds donnés et trouvent une représentation vectorielle pour chaque nœud. Comparé à d’autres types de données, la caractéristique la plus importante d’un graphique est qu’il peut clairement indiquer quels nœuds sont directement liés les uns aux autres et la proximité des relations entre les différents nœuds.Par conséquent, l’essence des réseaux neuronaux graphiques réside dans l’utilisation de ces biais inductifs et dans la transmission de messages entre les nœuds connectés. Plus les nœuds voisins sont proches, plus leur influence sur le nœud central est grande.
Ensuite, je partagerai avec vous plusieurs réseaux neuronaux convolutifs graphiques classiques.
Le premier est le réseau neuronal convolutif graphique GCN, Comme le montre la figure ci-dessous, l’essentiel est que chaque couche de GCN agrège en moyenne les informations des voisins du premier ordre au nœud central et utilise les informations agrégées comme une nouvelle représentation du nœud central.
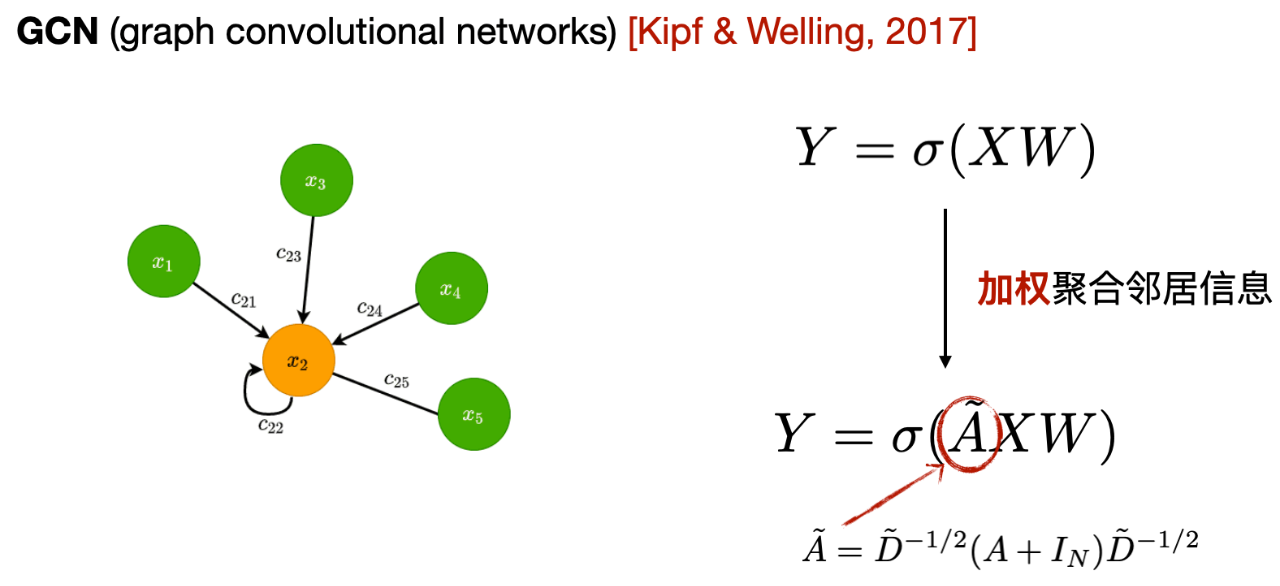
À partir de l'expression, nous pouvons voir que la différence entre GCN et MLP est que GCN ajoute la matrice d'adjacence et utilise les informations de voisinage du premier ordre pour mettre à jour la représentation du nœud. De plus, il ajoute des boucles automatiques pour renforcer ses propres informations lors de l'agrégation des informations et effectue une moyenne pondérée en fonction du nombre de voisins de chaque nœud voisin.
- Voisins du premier ordre : Le nœud central est directement connecté aux autres nœuds, c'est-à-dire que les points qui peuvent être atteints par une arête sont appelés voisins du premier ordre.
Le deuxième est le Graph Attention Network (GAT). Par rapport à GCN, le principal changement de GAT est la manière de calculer les poids lors de l'agrégation des informations voisines. GCN utilise des poids calculés en fonction de la matrice d'adjacence, tandis que GAT calcule un poids apprenable en fonction des caractéristiques des nœuds voisins.
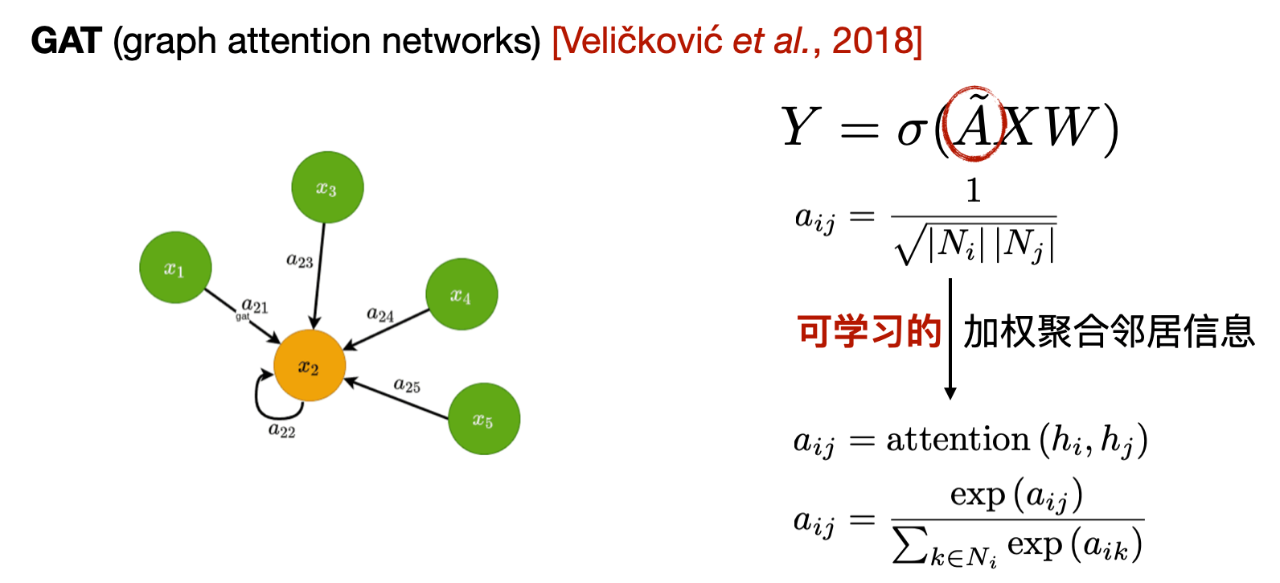
Les deux méthodes ci-dessus sont des représentants typiques des méthodes transductives. Ils nécessitent un graphique complet en entrée, ce qui augmente la complexité de calcul.À cet égard, GraphSAGE propose une approche inductive. Chaque fois que des informations sont transmises, il suffit de comprendre les voisins de premier ordre du nœud central, et seule une partie des informations sur les voisins est sélectionnée au hasard pour l'agrégation.
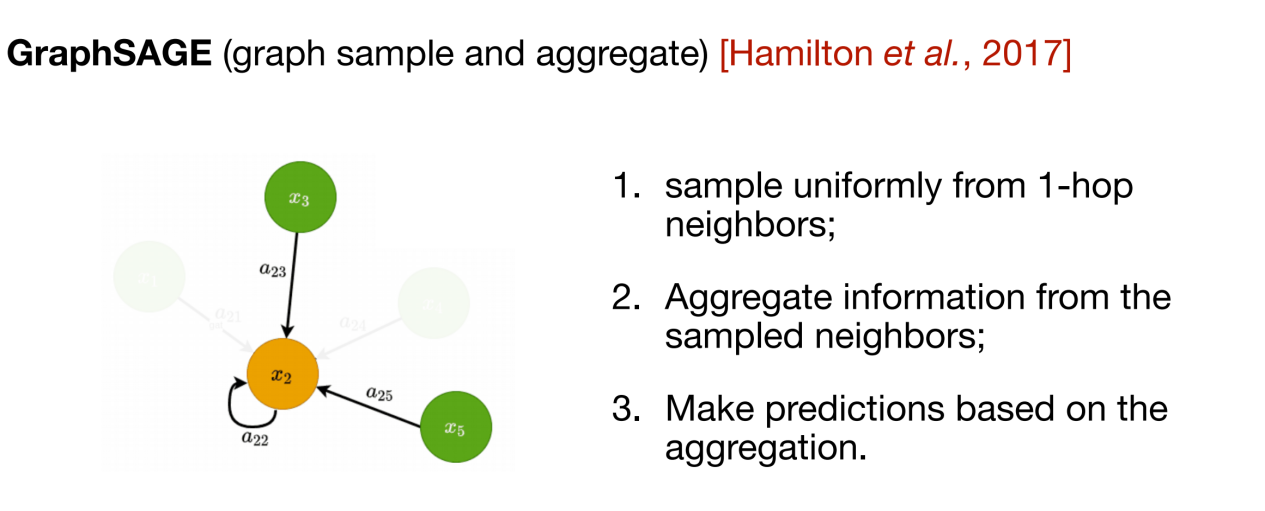
Les trois méthodes ci-dessus visent à mettre à jour la représentation des nœuds sur le graphe de structure topologique bidimensionnel, et le réseau de transmission de messages ultérieur (MPNN) intègre ce type de méthode d'agrégation d'informations dans un cadre. Cependant, de nombreuses données biologiques (telles que les molécules) doivent également prendre en compte la structure tridimensionnelle.Afin d'intégrer des informations spatiales, un réseau neuronal à graphes équivariants (EGNN) peut être utilisé. Comme le montre la figure ci-dessous, le cœur de cette méthode est qu’en plus des informations sur les caractéristiques du nœud lui-même, la relation de position relative entre les nœuds est également introduite pour garantir l’équivariance rotationnelle et l’invariance translationnelle de la représentation apprise.

En outre, il existe de nombreuses conceptions avancées de réseaux neuronaux graphiques. Certaines conceptions peuvent non seulement améliorer les performances prédictives du modèle, mais également se concentrer sur l'amélioration de l'efficacité, la réduction du lissage excessif, l'ajout d'une représentation multi-échelle et d'autres besoins. En introduisant le passage continu de messages, les méthodes de convolution de graphes spectraux, etc., il peut également fournir des réseaux neuronaux graphiques plus expressifs pour des problèmes spécifiques.
Applications importantes des réseaux neuronaux graphiques : prendre comme exemple la prédiction des propriétés des protéines et la génération de séquences
Ensuite, je partagerai avec vous l’application des réseaux neuronaux graphiques dans l’apprentissage de la représentation des protéines.Ici, je le divise en deux catégories : les modèles de prédiction et les modèles de génération.
Codage des caractéristiques des protéines et prédiction des propriétés
En termes de tâches de prédiction, nous considérons trois types de tâches : la prédiction des propriétés mutantes, la prédiction de la solubilité et la correspondance des sous-graphes, qui sont quatre tâches spécifiques.
Le premier travail porte sur la prédiction des tâches de mutation. Comme le montre la figure ci-dessous, nous avons utilisé des réseaux neuronaux à graphes équivariants pour caractériser les relations spatiales internes des acides aminés protéiques, où chaque nœud représente un acide aminé, indiquant le type, les propriétés physiques et chimiques et d'autres caractéristiques de l'acide aminé au point. Les connexions de bord sur le graphique reflètent les relations entre les acides aminés, telles que le potentiel d'évolution commune et l'influence des forces mutuelles.
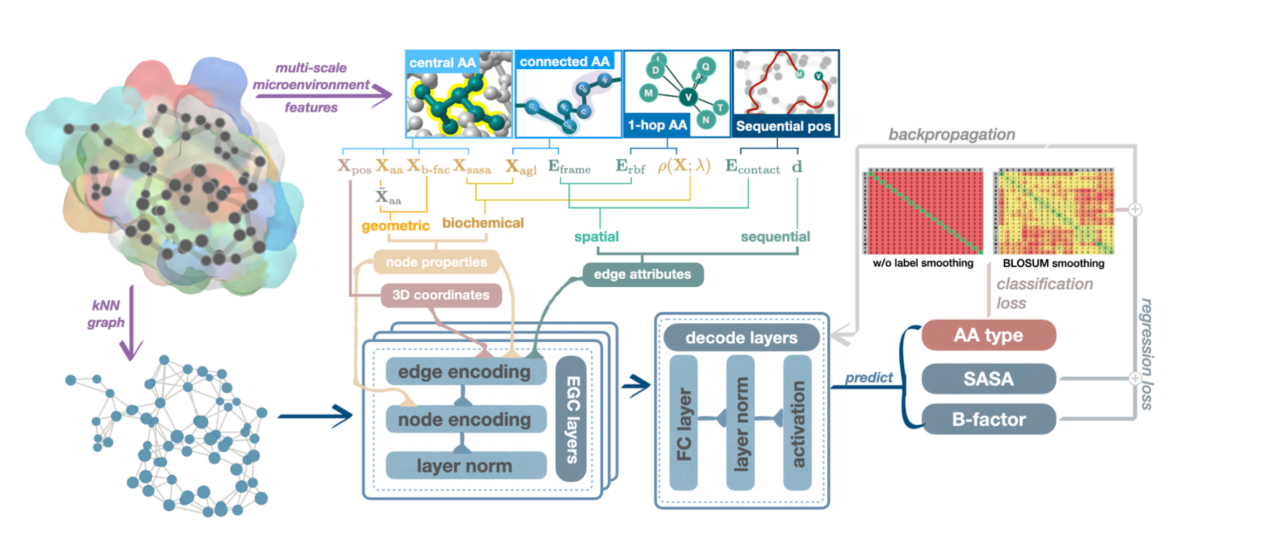
Nous avons ensuite utilisé un modèle prédictif pour évaluer différents mutants et identifier les combinaisons de mutations à score élevé qui sont les plus susceptibles d’optimiser les propriétés des protéines. Ce réseau neuronal graphique léger peut réduire considérablement les coûts de formation et de données en intégrant les acides aminés et les relations entre les acides aminés, rendant le modèle petit et beau tout en maintenant des performances élevées. De plus, la vérification par des expériences humides sur diverses propriétés des protéines a démontré que ce modèle peut améliorer considérablement l’effet et le taux de réussite de l’évolution dirigée. La recherche a été intitulée « Ingénierie des protéines avec des réseaux neuronaux légers de débruitage de graphes » et publiée dans ACS JCIM.
Adresse du document :
https://pubs.acs.org/doi/10.1021/acs.jcim.4c00036
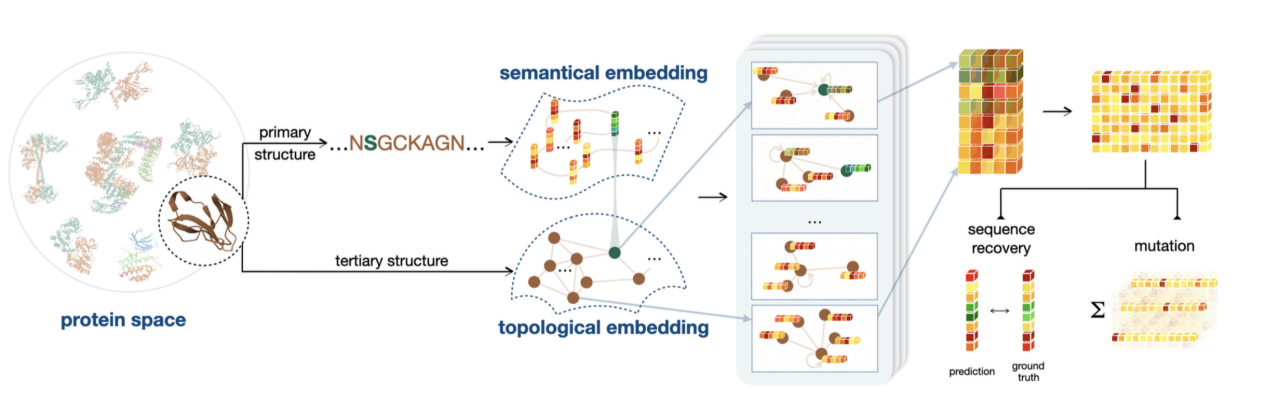
La deuxième tâche consiste à ajouter un codage de séquence protéique basé sur un codage structurel. Cela est dû au fait que les informations structurelles supposent que les interactions entre les acides aminés adjacents sont plus fortes, tandis que les interactions à longue distance sont extrêmement faibles. Cette hypothèse ne correspond pas entièrement à la situation réelle, des informations sur la séquence sont donc nécessaires pour compléter la prise en compte des interactions à longue portée. De plus, les informations sur les différentes propriétés biologiques ont des accents différents. Pour l’énergie de liaison et la stabilité thermique, les informations structurelles jouent un rôle dominant, mais lorsqu’il s’agit de propriétés telles que l’activité catalytique, les informations sur le type d’acides aminés sont plus critiques.
Comme le montre la figure ci-dessous, nous avons effectué des tests expérimentaux sur plus de 200 dosages sur ProteinGym et obtenu les meilleures performances des méthodes non MSA. L'étude a été publiée dans eLife sous le titre « Codage sémantique et géométrique des protéines vers une bioactivité et une thermostabilité améliorées ».
Adresse du document :
https://elifesciences.org/reviewed-preprints/98033
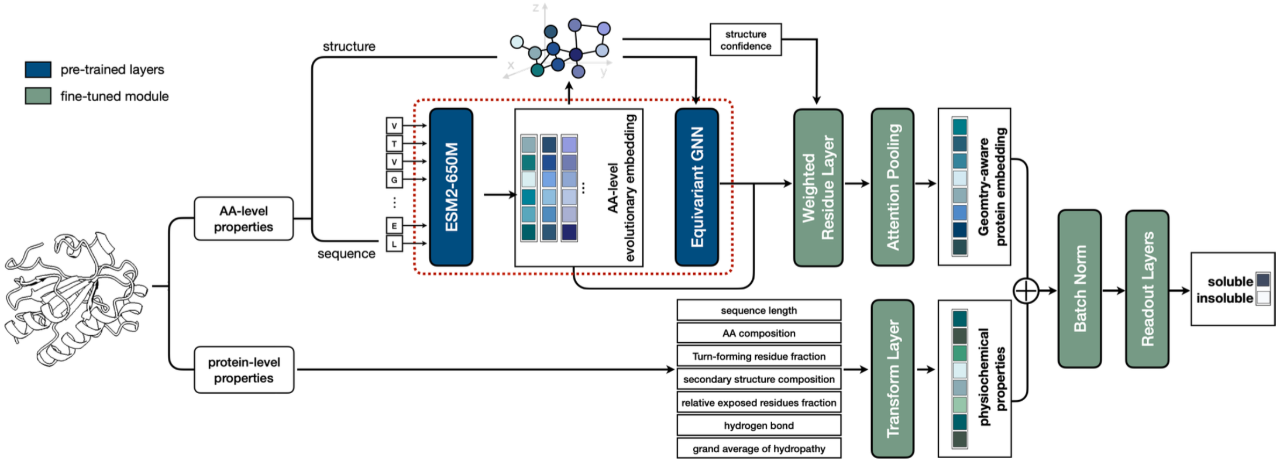
Le module de codage au niveau des acides aminés du troisième ouvrage est cohérent avec celui du deuxième ouvrage. L’intégration de l’information est basée sur la séquence et la structure des protéines. La différence est qu’il intègre également une variété d’informations au niveau des protéines basées sur des connaissances préalables, telles que la longueur des protéines, la distribution proportionnelle de 20 acides aminés, etc.
Comme le montre la figure ci-dessous, nous avons testé l’effet prédictif du modèle sur la solubilité des protéines et obtenu des résultats SOTA sur des milliers de données de test basées sur des calculs et des expériences. La recherche, intitulée « ProtSolM : prédiction de la solubilité des protéines avec des caractéristiques multimodales », a été acceptée par IEEE BIBM2024 (conférence CCF de catégorie B).
Adresse de l'article pré-imprimé :
https://www.arxiv.org/abs/2406.19744
La quatrième tâche consiste à explorer les similitudes locales des structures des protéines. Comme le montre la figure ci-dessous, bien que la protéine soit grande dans son ensemble, son noyau peut résider dans certaines caractéristiques structurelles locales. De plus, d’un point de vue macroscopique, deux protéines peuvent être complètement différentes en termes de séquence et de structure, mais avoir des modules fonctionnels de base similaires, voire identiques.
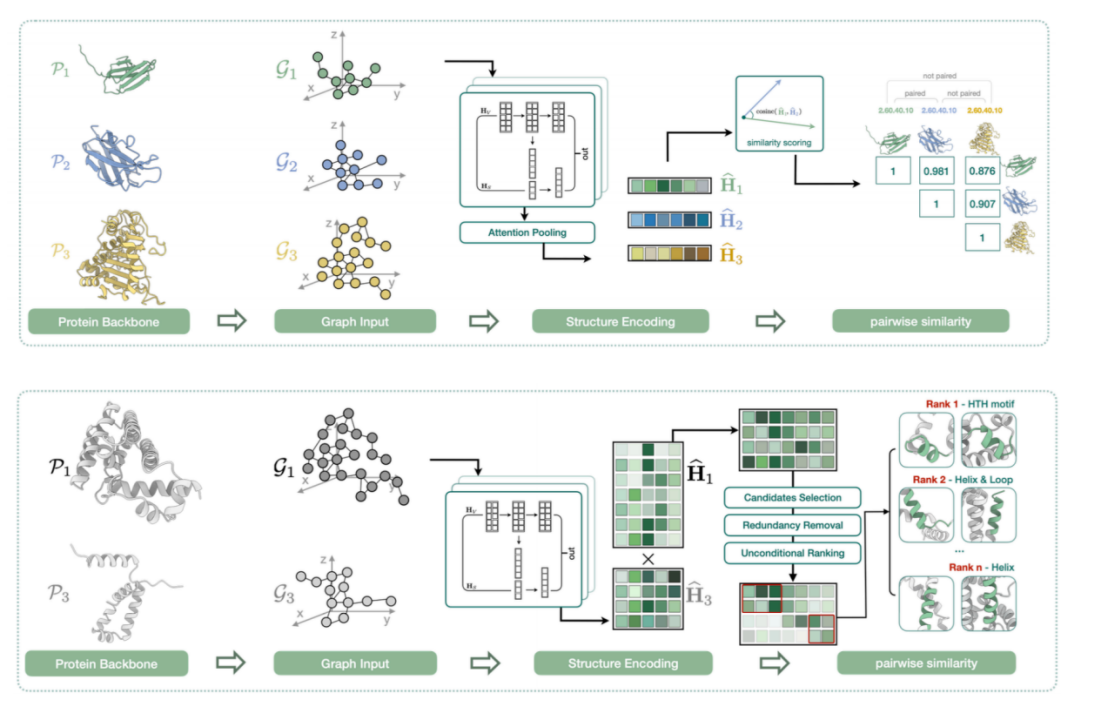
Nous trouvons donc une représentation implicite de la structure locale de chaque protéine et calculons la similarité entre ces vecteurs. En plus de comparer la similarité biunivoque entre les structures, nous évaluons également s'il existe des fragments de structure locale alignables entre 2 protéines complètes. La recherche, intitulée « Apprentissage de la représentation des protéines avec intégration des informations de séquence : conduit-elle toujours à de meilleures performances ? », a été acceptée par IEEE BIBM2024.
Adresse de l'article pré-imprimé :
https://arxiv.org/abs/2406.19755
Génération de séquences
Ensuite, je partagerai avec vous deux travaux, à savoir la conception de séquences d'acides aminés adaptées aux structures protéiques. Les modèles centraux de ces deux travaux sont des modèles de probabilité de diffusion (Diffusion).
La première tâche consiste à concevoir une séquence protéique complète basée sur le squelette d’acides aminés connu pour améliorer les performances des protéines. Le cadre du modèle est illustré dans la figure ci-dessous. Contrairement à l’évolution dirigée, nous avons modifié des centaines d’acides aminés à la fois et obtenu des séquences protéiques avec une plus grande diversité. D’une part, cette méthode peut trouver un point de départ complètement nouveau pour l’évolution et éviter des problèmes tels que l’optimalité locale et les effets négatifs à la hausse qui sont courants dans l’évolution dirigée ; d'autre part, en modifiant davantage d'acides aminés pour obtenir des protéines avec une similarité de séquence plus faible mais la même fonction, il devient possible de briser le blocage des brevets.
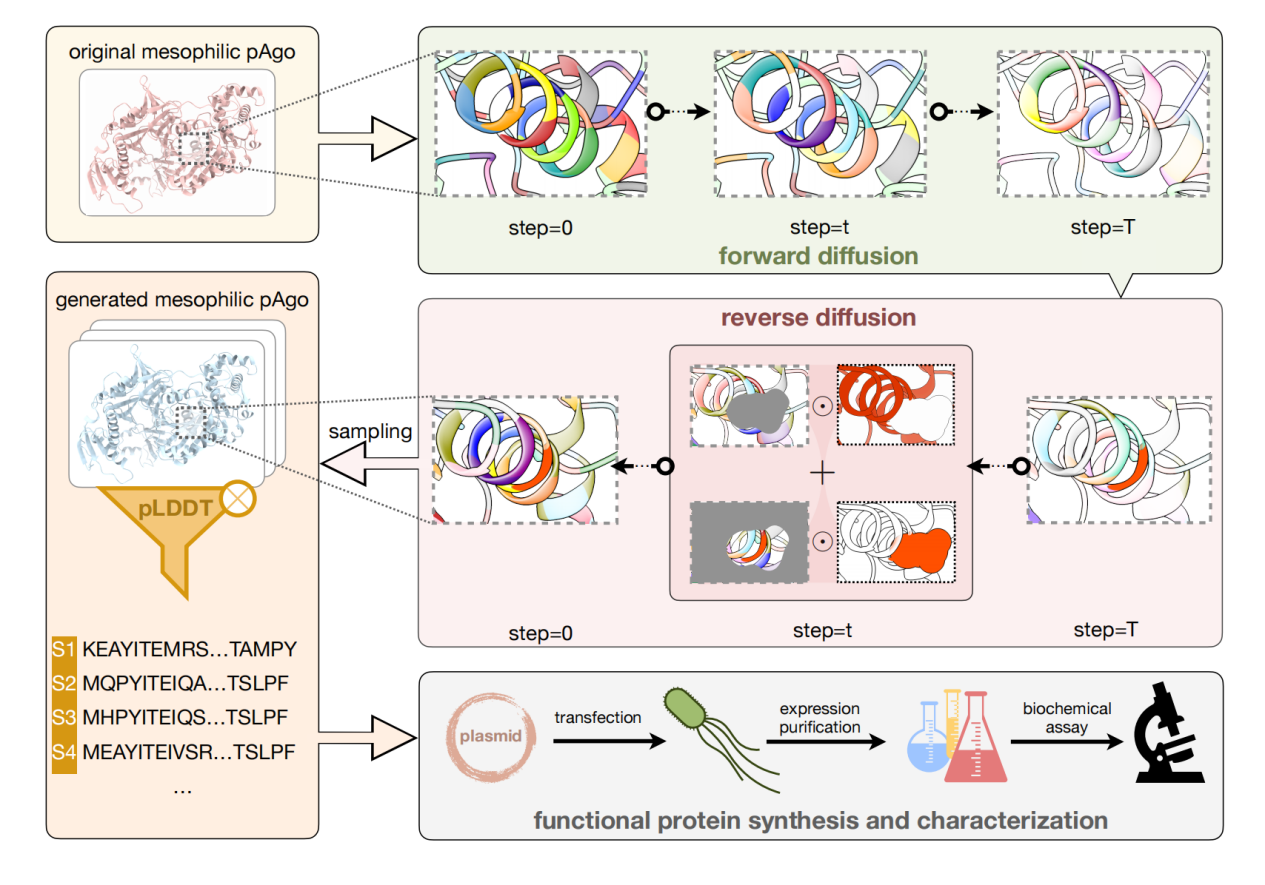
Nous avons utilisé deux protéines Argonaute (fonctionnant respectivement à des températures moyennes et ultra-élevées) comme modèles de conception, et la plupart des plus de 40 protéines générées peuvent effectuer le clivage de l'ADN à température ambiante. La meilleure conception présente une activité de clivage plus de 10 fois supérieure à celle du type sauvage, et sa stabilité thermique est également considérablement améliorée. L’étude, intitulée « La diffusion conditionnelle de débruitage des protéines génère des endonucléases programmables », a été publiée dans Cell Discovery.
Adresse de l'article pré-imprimé :
https://www.biorxiv.org/content/10.1101/2023.08.10.552783v1
La deuxième tâche, comme le montre la figure ci-dessous, consiste à déterminer indépendamment le nombre et la position des acides aminés à remplir en fonction de la structure secondaire sans restreindre strictement la structure du squelette des acides aminés. Par rapport à la méthode de génération basée sur le squelette, cette condition de génération plus grossière peut introduire une diversité de séquence dans la séquence générée et peut également répondre aux besoins spécifiques de modification des protéines et de nouvelle conception (comme pour les protéines transmembranaires, seule la partie transmembranaire est contrainte d'être une structure en hélice, mais la longueur et le squelette spécifique de cette partie ne sont pas strictement limités). L'étude, intitulée « Génération de nouvelles séquences de protéines guidées par la structure secondaire avec diffusion de graphes latents », a été acceptée par ICML AI4Science et le texte intégral est en cours de révision.
Adresse de l'article pré-imprimé :
https://arxiv.org/html/2407.07443v1
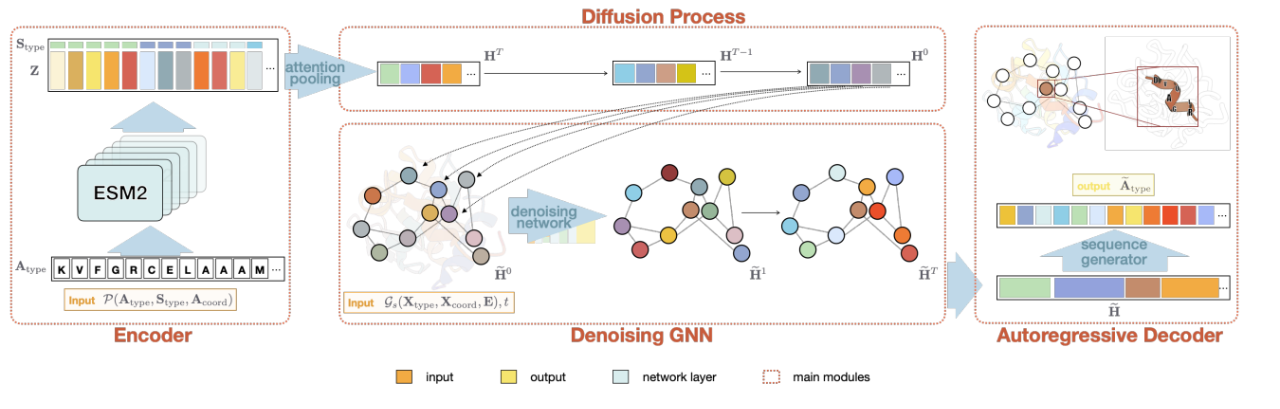
Les deux travaux de conception de séquences protéiques ci-dessus basés sur la diffusion peuvent soit générer une séquence entière selon le squelette protéique, soit fixer certains acides aminés clés et la structure du squelette et les utiliser comme conditions de génération pour remplir la séquence d'acides aminés de la partie non fixée.
Application des réseaux neuronaux graphiques à des problèmes plus biologiques
En plus de la modélisation conventionnelle des graphes moléculaires, les réseaux neuronaux graphiques peuvent également être appliqués à d'autres types de données et de problèmes pour promouvoir la recherche de problèmes plus biologiques. Ensuite, je vais partager deux exemples.
Le premier exemple est l’analyse et la simplification des réseaux sociaux biologiques. Tout comme les relations complexes dans les réseaux sociaux humains, il existe de nombreux contenus qui méritent d’être explorés dans les réseaux sociaux biologiques à différents niveaux (tels que les réseaux microbiens, les réseaux génétiques, etc.).
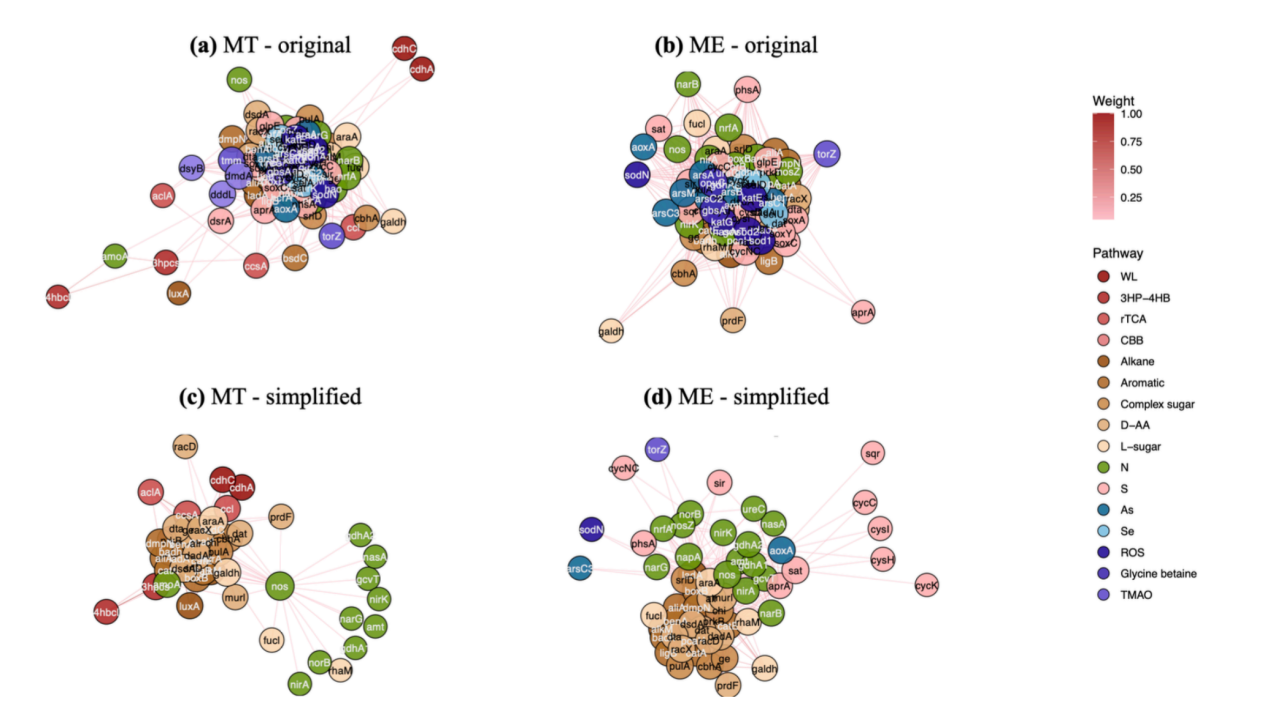
Nous avons déjà utilisé des réseaux de cooccurrence de gènes pour mener des études de simplification des réseaux sociaux. Comme le montre la figure ci-dessous, les figures a et b sont des réseaux différents du même gène provenant des profondeurs marines et des hautes montagnes. Leurs formes originales sont complexes et désorganisées. En construisant un réseau neuronal graphique similaire au réseau social humain, nous simplifions les deux réseaux, identifions les gènes qui occupent une position dominante absolue et distinguons quels gènes ont des connexions plus étroites et quels gènes ont des connexions relativement faibles. Le réseau simplifié peut aider les biologistes à utiliser leur expertise pour analyser les réseaux et les communautés biologiques. Une version préliminaire de l'étude s'intitule « Une vue unifiée sur la transmission de messages neuronaux avec la dynamique d'opinion pour les réseaux sociaux ».
Adresse de l'article pré-imprimé :
https://arxiv.org/abs/2310.01272
Le deuxième exemple est la recherche d’interprétabilité basée sur un réseau neuronal graphique. Un exemple intuitif est que les réseaux neuronaux graphiques peuvent aider à identifier les structures locales clés au sein des molécules. D’une part, ce résultat peut être utilisé pour tester la rationalité du modèle. Par exemple, lors de la prédiction de la fonction des protéines, si le modèle peut localiser des atomes clés ou des acides aminés à proximité du centre actif dans une certaine mesure, cela signifie que le modèle a une certaine rationalité. Au contraire, si l’attention du modèle est distribuée de manière aléatoire et discrète sur plusieurs acides aminés à la surface de la protéine, alors il peut y avoir des problèmes avec le modèle. D’un autre côté, idéalement, un modèle explicatif raisonnable et puissant, en analysant le rôle de chaque nœud dans la prédiction fonctionnelle, pourrait même aider à identifier des régions de poche de protéines complètement nouvelles à l’avenir.
Bien que les grands modèles aient fourni une riche expérience réussie dans de nombreux scénarios d’application, ils ne constituent pas la seule solution à tous les problèmes. En tant que domaine dans lequel diverses données structurées existent naturellement, les réseaux neuronaux graphiques peuvent fournir des solutions possibles à de nombreux problèmes en biologie. Qu'il s'agisse de molécules, de complexes, de gènes, de réseaux microbiens ou de systèmes plus grands et plus complexes, les réseaux neuronaux graphiques peuvent fournir une solution simple en implantant des biais inductifs et en maximisant les connaissances humaines préalables, même dans le cas de petites quantités de données.
À propos de Zhou Bingxin

Zhou Bingxin est actuellement chercheur adjoint au Centre national de mathématiques appliquées (Université Jiao Tong de Shanghai). Elle a obtenu son doctorat à l'Université de Sydney, en Australie, en 2022, et a été chercheuse invitée à l'Université de Cambridge, au Royaume-Uni. Ses recherches portent sur l’utilisation de l’apprentissage profond (en particulier l’apprentissage profond géométrique) pour résoudre des défis en biologie, tels que l’ingénierie enzymatique, les réseaux de gènes métaboliques et l’analyse de l’évolution des groupes de structures protéiques. Les algorithmes d'apprentissage profond développés sont utilisés pour traiter des graphes statiques, dynamiques, hétérogènes et bruyants, et certains d'entre eux ont été publiés dans des revues et conférences internationales de premier plan telles que IEEE TPAMI, JMLR, ICML et NeurIPS. Le cadre général d'apprentissage profond pour l'ingénierie des protéines et la conception de séquences peut concevoir efficacement et améliorer considérablement l'activité de protéines complexes, et certains des résultats ont été publiés dans des revues telles que eLife, Chem. Sci., et ACS JCIM.
Page d'accueil personnelle :
https://ins.sjtu.edu.cn/peoples/ZhouBingxin
Google Scholar:








