Command Palette
Search for a command to run...
Une Nouvelle Ère D’exploration Matérielle ! L'équipe De Xu Yong Et Duan Wenhui De l'Université Tsinghua a Publié Un Cadre Fonctionnel De Densité De Réseau Neuronal Pour Ouvrir La Boîte Noire De La Prédiction De La Structure Électronique !

La théorie de la fonctionnelle de la densité (DFT), en tant qu'outil clé pour prédire et expliquer les propriétés des matériaux, joue un rôle indispensable en physique, en chimie, en science des matériaux et dans d'autres domaines.Cependant, la DFT traditionnelle nécessite généralement beaucoup de ressources informatiques et de temps, et son champ d’application est considérablement limité.
En tant que domaine interdisciplinaire émergent, la combinaison de l'apprentissage profond avec la théorie fonctionnelle de la densité pour prédire et découvrir de nouveaux matériaux grâce à un grand nombre de simulations informatiques surmonte progressivement les lacunes des calculs DFT traditionnels qui sont longs et complexes, et présente un grand potentiel d'application dans la construction de bases de données de matériaux informatiques. Les algorithmes de réseaux neuronaux accélèrent la construction de bases de données de matériaux plus volumineuses, et des ensembles de données plus volumineux peuvent former des modèles de réseaux neuronaux plus puissants. Cependant, la plupart des recherches actuelles sur l’apprentissage profond DFT traitent la tâche DFT et le réseau neuronal séparément, ce qui limite considérablement le développement collaboratif entre les deux.
Afin de combiner de manière plus organique l'algorithme du réseau neuronal et l'algorithme DFT, le groupe de recherche de Xu Yong et Duan Wenhui de l'Université Tsinghua a proposé le cadre de la théorie fonctionnelle de la densité du réseau neuronal (DFT du réseau neuronal).Ce cadre unifie la minimisation des fonctions de perte dans les réseaux neuronaux et l'optimisation des fonctionnelles énergétiques dans la théorie de la fonctionnelle de la densité. Comparé aux méthodes d'apprentissage supervisé traditionnelles, il offre une précision et une efficacité supérieures et ouvre une nouvelle voie au développement de méthodes DFT d'apprentissage en profondeur.
La recherche s'intitule « Théorie de la fonctionnelle de la densité des réseaux neuronaux basée sur la minimisation variationnelle de l'énergie » et est publiée dans Phys. Rév. Lett.
Points saillants de la recherche :
* Cette étude a proposé un cadre théorique pour la théorie fonctionnelle de la densité des réseaux neuronaux, combinant la théorie fonctionnelle de la densité variationnelle avec un réseau neuronal équivalent
* Cette étude est basée sur le langage Julia et combinée avec le cadre de différenciation automatique Zygote pour développer un programme informatique appelé AI2DFT à partir de zéro. Dans AI2DFT, la différenciation automatique (AD) peut être utilisée à la fois pour les calculs DFT variationnels et pour l'entraînement des réseaux neuronaux.
Adresse du document :
https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.133.076401
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Apprentissage profond et intégration DFT pour réaliser un apprentissage non supervisé d'informations physiques
Kohn-Sham DFT est la méthode de calcul des premiers principes la plus utilisée dans la simulation des matériaux.Cette méthode simplifie le problème complexe d'interaction électronique en le transposant dans un problème électronique simplifié sans interaction décrit par un hamiltonien Kohn-Sham à particule unique efficace, et utilise une fonction d'échange-corrélation approximative pour prendre en compte les effets complexes à plusieurs corps. Bien que l'équation de Kohn-Sham soit formellement dérivée du principe variationnel et soit très populaire dans le domaine de la physique théorique, elle n'est pas souvent utilisée dans les calculs DFT car les solutions itératives des équations différentielles sont plus efficaces.
Cependant, avec l’intégration plus poussée de l’apprentissage profond et de la DFT, ce paradigme de développement a été complètement changé.
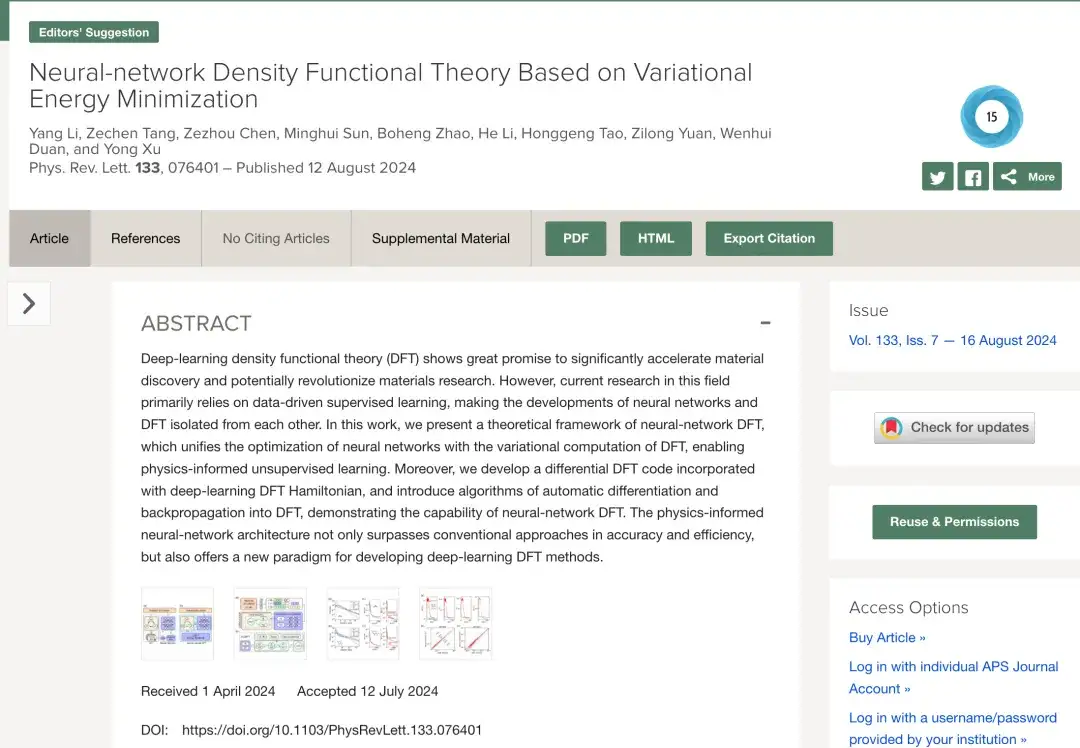
La recherche sur l'apprentissage profond DFT s'appuie principalement sur des techniques d'apprentissage supervisé basées sur les données. Comme le montre la figure a ci-dessous, en général, les méthodes traditionnelles d'apprentissage supervisé basées sur les données génèrent des données de formation en effectuant des calculs DFT SCF sur différentes structures matérielles, puis conçoivent et forment un réseau neuronal pour prédire des données similaires aux résultats DFT. Dans ce processus, le réseau neuronal et la DFT sont séparés.
De nos jours, comme le montre la figure b ci-dessus, l'apprentissage non supervisé d'informations physiques basé sur le réseau neuronal DFT réalise le partage et la collaboration de l'algorithme du réseau neuronal et de l'algorithme DFT en combinant l'optimisation de la fonctionnelle énergétique dans le DFT avec la minimisation de la fonction de perte dans le réseau neuronal.Plus important encore, en introduisant explicitement la DFT dans l’apprentissage profond, le modèle de réseau neuronal peut simuler l’environnement physique réel mieux que les méthodes d’entraînement des données précédentes.
L'introduction de techniques de différenciation automatique et de rétropropagation implémente numériquement le réseau neuronal DFT
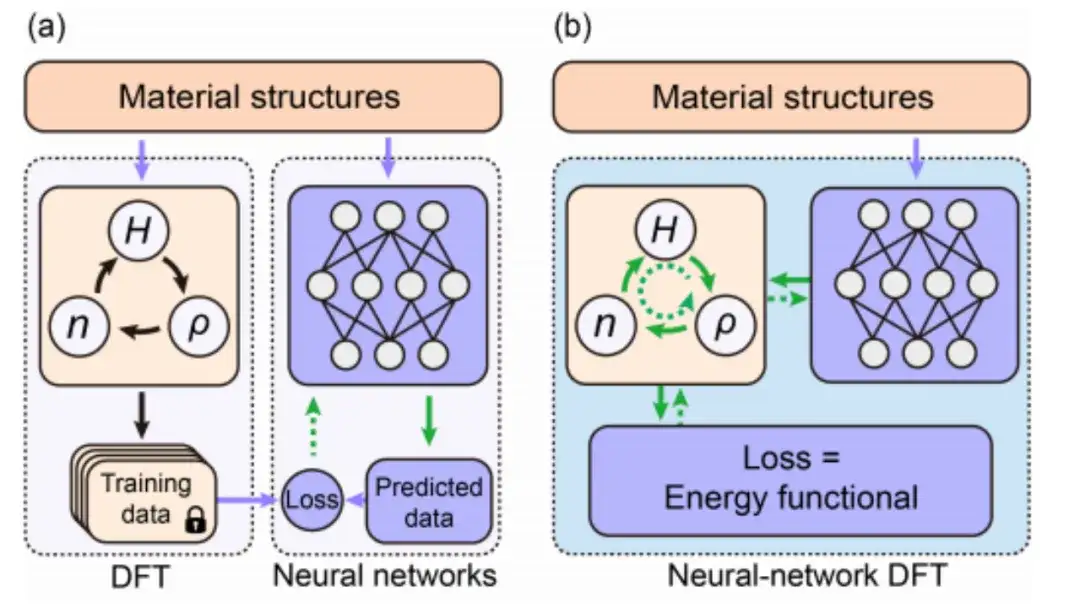
Cette étude a sélectionné la fonctionnelle énergétique E[H] pour une étude de suivi et a adopté DeepH-E3 pour assurer la covariance de la cartographie {R} → H dans le groupe euclidien tridimensionnel.
* DeepH-E3, l'architecture DeepH de deuxième génération conçue par l'équipe de Xu Yong sur la base de réseaux neuronaux équivalents. Ce cadre utilise un réseau neuronal covariant sous un groupe euclidien tridimensionnel pour prédire l'hamiltonien de la théorie fonctionnelle de la densité (DFT) correspondant à la structure atomique microscopique, accélérant considérablement le calcul de la structure électronique des premiers principes.
Plus précisément, comme le montre la figure ci-dessous, cette étude utilise l'intégration des informations sur la structure matérielle comme condition d'entrée du réseau neuronal équivariant, puis génère la matrice hamiltonienne pour obtenir le poids du réseau neuronal paramétré Hθ. La fonctionnelle énergétique E[H] peut également être considérée comme la fonction de perte E[Hθ] du réseau neuronal.
Dans la DFT du réseau neuronal, le programme DFT doit fournir ∇HE pour optimiser les paramètres du réseau neuronal, ce qui constitue un défi majeur pour la programmation DFT.
Bien que la différenciation automatique (AD) soit bien adaptée au calcul de ∇HE, la plupart des codes DFT actuels ne prennent pas entièrement en charge la fonctionnalité AD. Par conséquent, cette étude a développé un programme DFT différentiable automatique autonome et utilisable « AI2DFT » utilisant le langage Julia. Il convient de noter que dans AI2DFT, la différenciation automatique (AD) est appliquée à la fois au calcul DFT et à la formation du réseau neuronal.
Dans DFT, AI2DFT peut déduire ρ et n de H pour calculer l'énergie totale, puis utiliser le mode inverse AD pour calculer ∇nE, ∇ρE et ∇HE en séquence en fonction de la règle de la chaîne. Dans les réseaux neuronaux, les informations de gradient ∇θE peuvent être utilisées pour l'optimisation du réseau neuronal.
Résultats de la recherche : Le réseau neuronal DFT présente une grande fiabilité et d'excellentes performances en termes de précision de prédiction
Après avoir établi le cadre théorique du réseau neuronal DFT et terminé l'implémentation numérique à l'aide du code différentiable AI2DFT, l'étude a mené des tests complets sur une série de matériaux comprenant des molécules H2O, du graphène, du MoS2 monocouche et du Na cubique centré.
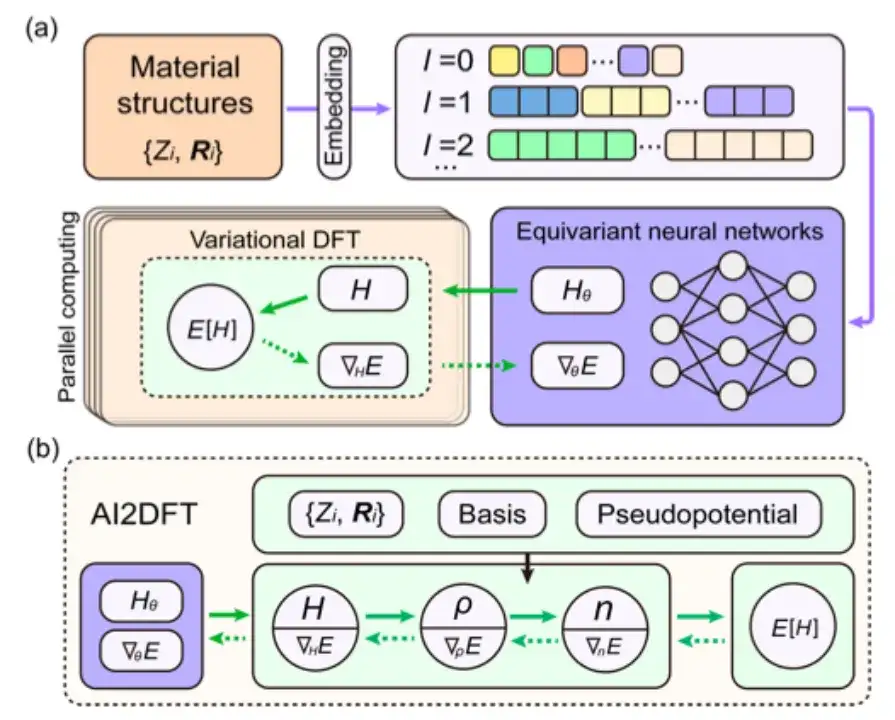
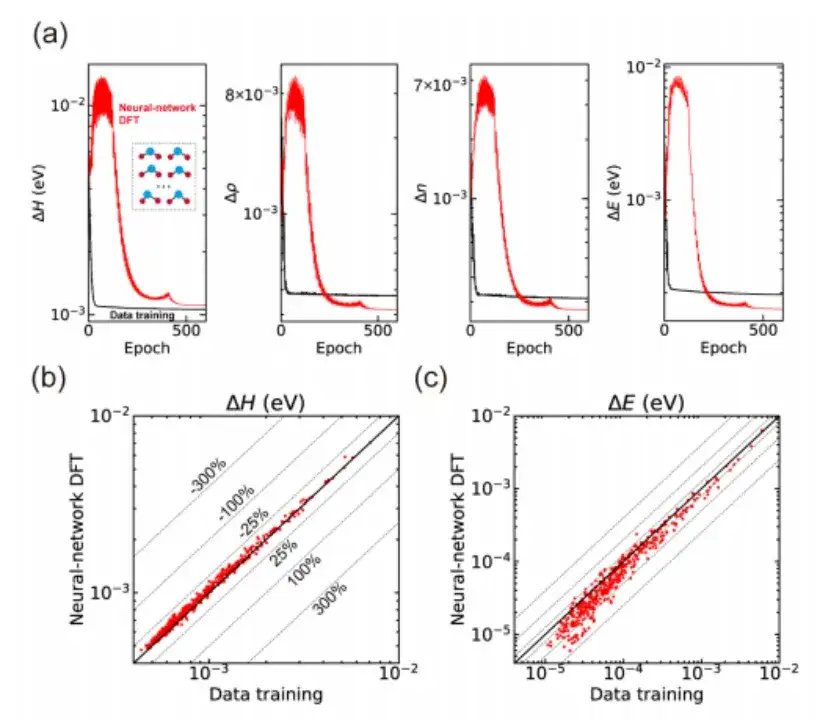
Plus précisément, l’étude a d’abord vérifié si l’itération SCF d’AI2DFT pouvait bien reproduire les résultats de référence du code SIESTA, puis a appliqué la DFT variationnelle pour étudier les mêmes matériaux.Comme le montrent les figures a et b ci-dessous, après des dizaines d'étapes variationnelles, l'énergie totale peut converger en dessous de l'échelle μeV, et d'autres quantités physiques telles que le gradient d'énergie, l'hamiltonien, la matrice de densité et la densité de charge montrent également une convergence exponentielle, ce qui vérifie la fiabilité et la robustesse de la DFT variationnelle.
De plus, comme le montrent les figures c et d ci-dessus, par rapport aux méthodes traditionnelles d’apprentissage supervisé basées sur les données, AI2DFT combine les performances de la DFT variationnelle et du réseau neuronal DeepH-E3. Par exemple:
* Pour la molécule H2O, l'hamiltonien DFT peut être optimisé par le réseau neuronal DFT pour atteindre une grande précision, avec une précision de 0,06 meV pour le réseau neuronal DFT et de 0,02 meV pour l'apprentissage des données.
* Pour le graphène, les deux méthodes ont atteint une précision supérieure de 0,004 meV. Par conséquent, la fiabilité de la méthode du réseau neuronal a été vérifiée.
* De plus, en termes de précision de prédiction énergétique, les performances du réseau neuronal DFT sont nettement meilleures que celles de l'entraînement des données. Par exemple, pour la molécule H2O, la précision de prédiction d'énergie du réseau neuronal DFT atteint 0,013 μeV, ce qui est plus de 60 fois supérieur aux 0,83 μeV de l'entraînement des données.
Enfin, l’étude a également appliqué la DFT du réseau neuronal au calcul de diverses structures matérielles et a démontré la capacité d’apprentissage non supervisé.En prenant la molécule H2O comme exemple, comme le montre la figure a ci-dessous, l'étude a d'abord obtenu un modèle de réseau neuronal pré-entraîné grâce à un apprentissage supervisé basé sur les données basé sur la méthode DeepH-E3, puis a utilisé le réseau neuronal DFT pour affiner 300 structures d'entraînement afin d'obtenir des prédictions de haute précision de l'hamiltonien et d'autres quantités physiques.
De plus, l’étude a également utilisé le modèle de réseau neuronal formé pour prédire 435 structures de test qui n’avaient pas été vues pendant le processus de formation, et les résultats ont également montré une bonne capacité de généralisation, comme le montrent les figures b et c ci-dessus.Bien que l'hamiltonien produit par le DFT du réseau neuronal ait une erreur absolue moyenne légèrement plus grande que l'apprentissage supervisé basé sur les données, il montre des performances supérieures en termes de précision de prédiction, ce qui implique que le réseau neuronal apprend davantage de modèles physiques grâce au processus DFT du réseau neuronal.
En se concentrant sur la DFT et les calculs de premiers principes, l'IA ouvrira une nouvelle ère de la science des matériaux
Dirigée par le professeur Xu Yong et le professeur Duan Wenhui de l'Université Tsinghua, l'équipe de recherche a réuni Li Yang, Tang Zechen, Chen Zezhou et d'autres. Ils ont obtenu une série de résultats en théorie de la fonctionnelle de la densité (DFT) et en calculs de premiers principes. Leurs travaux ont favorisé l’application généralisée de la technologie d’apprentissage profond dans la recherche en science des matériaux et en physique.
Depuis 2022, l'équipe de recherche de Xu Yong et Duan Wenhui a réalisé des avancées majeures dans le domaine de l'informatique de base.Ils ont développé un cadre théorique et un algorithme basé sur l'apprentissage profond - DeepH (Deep DFT Hamiltonian).Cette méthode utilise pleinement le principe de localité des propriétés électroniques. Il suffit d'utiliser un modèle formé avec un petit ensemble de données système pour fournir des prédictions précises dans des systèmes de matériaux à grande échelle, améliorant considérablement l'efficacité de calcul de la structure électronique des matériaux non magnétiques.
Les résultats correspondants ont été publiés dans Nature Computational Science sous le titre « Théorie de la fonctionnelle de la densité par apprentissage profond Hamiltonien pour un calcul efficace de la structure électronique ab initio ».
Lien vers l'article :
https://www.nature.com/articles/s43588-022-00265-6
En 2023, afin d'étudier plus en détail la dépendance de l'hamiltonien DFT des matériaux magnétiques sur les atomes et les structures magnétiques, l'équipe de recherche de Xu Yong et Duan Wenhui a proposé la méthode xDeepH (DeepH étendu).Cette méthode utilise un cadre de réseau neuronal équivariant profond pour représenter l'hamiltonien DFT des matériaux magnétiques, effectuant ainsi des calculs de structure électronique efficaces. Cette réalisation fournit non seulement un outil de calcul efficace et précis pour l’étude des structures magnétiques, mais propose également une voie réalisable pour équilibrer la précision et l’efficacité de la DFT.
Les résultats correspondants ont été publiés dans Nature Computational Science sous le titre « Calcul de la structure électronique par apprentissage profond des superstructures magnétiques ».
Lien vers l'article :
https://www.nature.com/articles/s43588-023-00424-3
Afin de concevoir des modèles de réseaux neuronaux qui incluent des connaissances préalables et des exigences de symétrie, les chercheurs ont également proposé la méthode DeepH-E3.Cette méthode peut utiliser une petite quantité de données DFT pour former des systèmes à petite échelle, puis prédire rapidement la structure électronique des systèmes matériels à grande échelle. Il améliore non seulement considérablement la vitesse de calcul de plusieurs ordres de grandeur, mais atteint également un niveau élevé de précision de prédiction de l'ordre du sous-milliélectronvolt.
Les résultats associés ont été publiés dans Nature Communications sous le titre « Cadre général pour la représentation du réseau neuronal E(3)-équivariant de la théorie de la fonctionnelle de la densité hamiltonienne ».
Lien vers l'article :
https://www.nature.com/articles/s41467-023-38468-8
Grâce à l'optimisation continue du cadre théorique, DeepH a obtenu une couverture complète des matériaux non magnétiques aux matériaux magnétiques, et a également obtenu des améliorations significatives de la précision des prédictions.Dans ce contexte, l'équipe de recherche de Xu Yong et Duan Wenhui a également utilisé la méthode DeepH pour construire un modèle de matériau général DeepH.Le modèle est capable de gérer des systèmes de matériaux contenant plusieurs éléments et des structures atomiques complexes, et démontre une excellente précision dans la prédiction des propriétés des matériaux.
Les résultats de recherche pertinents ont été publiés dans Science Bulletin sous le titre « Modèle de matériaux universels de la théorie fonctionnelle de la densité d'apprentissage profond Hamiltonien ».
Lien vers l'article :
https://doi.org/10.1016/j.scib.2024.06.011
Aujourd'hui, le groupe de recherche de Xu Yong et Duan Wenhui a combiné de manière organique l'algorithme du réseau neuronal et l'algorithme DFT, ouvrant une nouvelle voie pour le développement de méthodes DFT d'apprentissage en profondeur. Il ne fait aucun doute qu’avec l’avancement continu des algorithmes de réseaux neuronaux profonds et l’établissement d’ensembles de données plus vastes, l’IA deviendra plus intelligente, et il est probable que dans un avenir proche, les calculs de premiers principes, la découverte de matériaux et la conception seront tous effectués par des réseaux neuronaux. Cela indique que l’IA mènera la science des matériaux vers une nouvelle ère axée sur les données.








