Command Palette
Search for a command to run...
Sélectionné Pour l'ICML ! Une Équipe Du MIT Réalise Une Nouvelle Avancée Basée Sur AlphaFold, Révélant La Diversité Dynamique Des Protéines
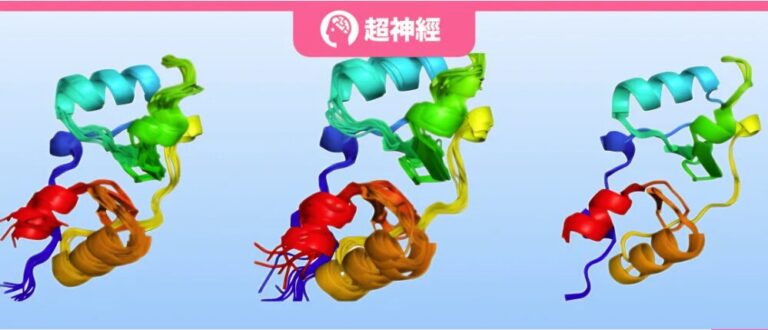
En tant que composant important des organismes, les protéines ont différents états et adoptent des structures tridimensionnelles complexes basées sur différentes combinaisons structurelles de mouvement collectif ou de fluctuations désordonnées pour exécuter de riches fonctions biologiques. Par exemple, les changements conformationnels des protéines sont essentiels aux fonctions des transporteurs, des canaux et des enzymes, tandis que les propriétés de la combinaison équilibrée aident à contrôler la force et la sélectivité des interactions moléculaires.
Ces dernières années, les méthodes d’apprentissage en profondeur telles qu’AlphaFold ont obtenu un grand succès dans la modélisation à état unique des protéines, mais ne peuvent pas expliquer l’hétérogénéité conformationnelle. Donc, pour les biologistes structuraux,Comment garantir une prédiction précise d’une structure unique tout en révélant des combinaisons structurelles potentielles ?C’est un problème difficile qui doit être résolu de toute urgence.
Récemment, une équipe de recherche du MIT a combiné les nouvelles méthodes d'échantillonnage d'AlphaFold et d'ESMFold et a fourni une nouvelle perspective pour observer et comprendre l'espace conformationnel des protéines grâce à la technologie de correspondance de flux.
Cette étude démontre les performances des variantes de correspondance de flux AlphaFlow et ESMFlow dans deux scénarios différents.Le modèle a finalement été affiné sur le PDB et formé davantage sur l'ensemble de données ATLAS, tous deux montrant des performances supérieures, dépassant non seulement la base de référence MSA traditionnelle dans la prédiction de la flexibilité conformationnelle et la modélisation de la distribution de position atomique, mais réalisant également des progrès significatifs dans la réplication des observations de groupes d'ordre supérieur.
La recherche connexe, intitulée « AlphaFold Meets Flow Matching for Generating Protein Ensembles », a été sélectionnée pour ICML 2024, la principale conférence universitaire dans le domaine de l'IA.

Adresse du document :
https://openreview.net/forum?id=rs8Sh2UASt
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : basé sur les ensembles de données PDB et ATLAS pour garantir l'équité des résultats expérimentaux
Comme nous le savons tous, AlphaFold a été développé et formé de manière de bout en bout sur la base des structures du PDB, tandis qu'ESMFold a utilisé des intégrations du Protein Language Model (PLM) comme entrée. donc,Cette étude a principalement utilisé l’ensemble de données PDB et l’ensemble de données MD.
Tout d'abord, pour construire un ensemble de tests de protéines structurellement hétérogènes à partir de la PDB, nous avons utilisé la base de données d'annotations SIFTS et sa cartographie au niveau des résidus des chaînes PDB aux séquences de référence UniProt, en associant chaque chaîne déposée à un fragment. Par la suite, tous les fragments des clusters ont été entièrement connectés sur la base d'un seuil de similarité Jaccard de 0,75, traitant chaque cluster résultant comme une protéine unique.Cela a donné lieu à 75 000 protéines.
En outre, l’étude a recueilli :
* Protéines qui n'ont pas soumis de chaînes avant la date limite de formation AlphaFold, mais qui ont déposé 2 à 30 chaînes après la date limite ;
* Protéines dont la longueur est comprise entre 256 et 768 résidus ;
* Protéines avec au moins 2 clusters structurels lorsque le seuil de regroupement de chaînes était de 0,85 lDDT-Cα symétrique et connectivité complète.
Finalement, 563 protéines représentées par 2 843 chaînes ont été obtenues.Les chercheurs ont extrait 100 protéines représentées par 500 chaînes pour former un ensemble de tests.
Deuxièmement, les chercheurs ont construit l’ensemble de données ATLAS sur la base de l’ensemble de données MD.Ce dernier était composé de 1 390 protéines sélectionnées sur la base de la classification du domaine ECOD.Pour chaque protéine, l'ensemble de données fournit trois simulations répétées de 100 ns de longueur, chaque simulation contenant 10 000 images. Pour former et valider ces trajectoires, nous avons d'abord généré des MSA pour les 1 390 entrées ATLAS en utilisant les séquences fournies et le pipeline ColabFold MMSeqs2.
Par la suite, les chercheurs ont sélectionné au hasard 300 conformations du pipeline de formation, en utilisant respectivement le 1er mai 2018 et le 1er mai 2019 comme dates limites de formation et de validation, et ont finalement obtenu 1265/39/82 ensembles de formation, de validation et de test.

Création de modèles : utilisation d'AlphaFold comme modèle de débruitage pour effectuer une correspondance de flux sur des collections de protéines
Compte tenu des défis considérables que représente le redéveloppement d’un modèle de distribution avec la même précision et les mêmes capacités de généralisation qu’AlphaFold, cette étude s’appuie sur les avancées conceptuelles récentes dans les modèles génératifs.Il est presque simple de réutiliser AlphaFold comme modèle génératif.

À ce jour, les architectures de modèles de diffusion typiques du texte vers les images modélisent presque toutes la distribution conditionnelle p(x | s) d'une image x conditionnée par un indice textuel s. Au cœur de ces modèles se trouve un réseau neuronal de débruitage qui prend en compte une image bruyante et une invite de texte et prédit une image propre.
Sur la base de ces conditions, ces modèles sont généralement formés à l’aide d’un objectif simple d’erreur quadratique moyenne (MSE). De même, un prédicteur de structure protéique formé avec une fonction de perte de type régression telle qu'AlphaFold ou ESMFold peut être converti en un modèle débruité simplement en fournissant une entrée de structure bruyante supplémentaire. Grâce à ces ajustements architecturaux, cette étude peut insérer davantage AlphaFold et ESMFold dans n’importe quel cadre de modélisation générative basé sur la débruitage itératif.
Cette étude estime que la conception du cadre de génération de correspondance de flux équivaut à sélectionner un chemin de probabilité conditionnelle pt(x | x1) et son champ vectoriel correspondant ut(x | x1). Par conséquent, cette étude définit un réseau neuronal reparamétré x1(x, t; θ) en échantillonnant le bruit x0 à partir de q(x0) et en l'interpolant linéairement avec le point de données x1 pour définir le chemin de probabilité conditionnelle.Ainsi, l’architecture AlphaFold est utilisée comme modèle de débruitage.
Pour appliquer la correspondance de flux aux structures protéiques, l'étude décrit également la structure par les coordonnées 3D de ses β-carbones (α-carbone pour la glycine) : x ∈ R^N×3. Cela garantit également que l’entrée du réseau neuronal est toujours une structure tridimensionnelle physiquement plausible, semblable à un polymère.
Étant donné que le cadre de correspondance de flux implique la définition et l’inversion des processus de bruit, il présente de nombreuses similitudes avec la diffusion harmonique des structures protéiques, qui convergent toutes deux vers la même distribution antérieure. Cependant, dans un cadre plus général,La correspondance de flux offre deux avantages principaux :
d'abord,La diffusion harmonique converge vers la distribution a priori uniquement dans la limite de temps infinie, et le taux de convergence dépend de la dimensionnalité des données, c'est-à-dire de la taille des protéines. Cela conduit à un changement de distribution au moment de l'inférence lorsque l'entraînement se fait uniquement sur des cultures de taille relativement petite.
Deuxièmement,La correspondance de flux fournit un moyen simple de gérer les résidus manquants très courants dans la PDB en les omettant simplement. En revanche, la diffusion harmonique crée des dépendances entre les positions atomiques, nécessitant une interpolation des données pour les résidus manquants.

Enfin, l'étude a affiné tous les poids d'AlphaFold et d'ESMFold sur PDB en fonction du cadre de correspondance des processus, et les dates limites de formation d'AlphaFold et d'ESMFold utilisées étaient respectivement le 1er mai 2018 et le 1er mai 2020. À la fin de cette phase de formation, l'étude a obtenu des variantes de correspondance de flux d'AlphaFold et d'ESMFold,Et je l'ai appelé AlphaFLOW et ESMFLOW.
Pour évaluer la capacité d'apprentissage à partir d'ensembles MD, les deux modèles ont été affinés sur l'ensemble de données ATLAS contenant des simulations MD entièrement atomiques. Après une formation avec respectivement 43 000 et 27 000 exemples supplémentaires,L’étude a obtenu des variantes de modèles spécifiques à MD - AlphaFLOW-MD et ESMFLOW-MD.
Résultats expérimentaux : Les performances dépassent celles des méthodes traditionnelles et offrent de larges perspectives d'application dans le domaine de la biologie structurale
Les chercheurs ont d’abord évalué les capacités d’AlphaFLOW et d’ESMFLOW pour diverses conformations de protéines déposées dans le PDB.
À cette fin, l'étude a construit un ensemble de tests contenant 100 protéines présentant des preuves de chaînes multiples et d'hétérogénéité conformationnelle déposées après la date limite de formation AlphaFold (1er mai 2018), et les a évaluées selon trois indicateurs majeurs : précision, rappel et diversité.

Les résultats montrent qu'AlphaFLOW est similaire au sous-échantillonnage MSA dans la mesure où les deux augmentent la diversité des prédictions au détriment de la précision, mais par rapport au sous-échantillonnage MSA, les variantes d'AlphaFLOW suivent nettement mieux les fronts de Pareto.
En termes de précision et de rappel,AlphaFLOW présente un comportement très similaire au sous-échantillonnage MSA.De manière assez surprenante, aucune des deux approches n’améliore significativement le rappel global par rapport à l’AlphaFold de base.
Dans l’ensemble, la précision d’ESMFold et d’ESMFLOW est relativement inférieure à celle de la famille de méthodes AlphaFold. Cependant, ESMFLOW est capable d’injecter beaucoup de diversité par rapport à l’ESMFold de base.Et améliorez la mémorisation sans pratiquement aucun sacrifice en termes de précision.
De plus, l’analyse RMWD de cette étude a montré qu’AlphaFlow était légèrement meilleur qu’AlphaFold pour prédire la position moyenne des atomes et significativement meilleur que le sous-échantillonnage MSA en termes de variance de modélisation.

Cette étude a également évalué la capacité d’AlphaFLOW et d’ESMFLOW à générer des ensembles MD proxy pour un ensemble de tests de 82 protéines dans la base de données ATLAS. Cette étude a utilisé des échantillons distincts de chaque méthode et a examiné la similitude de l’échantillon avec la population MD à l’aide d’une série d’évaluations.
Les résultats montrent queAlphaFLOW-MD permet d’obtenir des améliorations significatives en termes de similarité, dépassant de loin les performances du sous-échantillonnage MSA.

Étant donné que le MD est considéré comme la vraie valeur, il est coûteux de l’exécuter jusqu’à la convergence. Par conséquent, cette étude analyse plus en détail si AlphaFLOW peut fournir de meilleurs résultats avec un budget de calcul limité équivalent, par exemple en heures GPU. À cette fin, l’étude a réduit le nombre d’échantillons extraits d’AlphaFLOW (de 250 à 4) et raccourci la longueur de la trajectoire MD (de 100 ns à 160 ps).
Les résultats montrent que la qualité de l’ensemble AlphaFLOW reste constante, mais les trajectoires MD mettent plus de temps à atteindre ou à dépasser le même niveau de qualité.
Trois grands modèles généraux de pré-entraînement des protéines se distinguent et le domaine de la biologie structurale est plein de vitalité
Au cours des dernières années, les protéines et l’IA sont entrées en collision en permanence pour créer de nouvelles étincelles.À l’heure actuelle, la préformation universelle des protéines a formé une nouvelle situation de trois piliers.C'est-à-dire la série DeepMind Alphafold, la série RoseTTAFold de David Baker et la série Meta ESM. Sur la base de ces trois modèles, les résultats de recherches scientifiques pertinentes ont commencé à exploser. Au cours du seul premier semestre 2024, plusieurs résultats de recherche ont été publiés dans des revues de premier plan telles que Nature et Science.
En mars 2024, des chercheurs de la faculté de médecine de l'université de Caroline du Nord, de l'université de Californie à San Francisco, de l'université de Stanford et de l'université de Harvard ont publié une étude dans Science confirmant queLes structures prédites par AlphaFold2 peuvent guider la découverte future de médicaments.L'équipe de recherche a découvert qu'AlphaFold2 présentait une praticité significative en biologie structurale, conception de protéines, interactions, prédiction de cibles, prédiction de fonctions et mécanismes biologiques, et était capable de rechercher de nouveaux médicaments potentiels en examinant des milliards de composés et en faisant correspondre des bibliothèques avec des structures protéiques.
En mai 2024, l'équipe Google DeepMind a publié AlphaFold 3 dans Nature, étendant la technologie au-delà du repliement des protéines et prédisant avec précision la structure et les interactions des molécules vivantes telles que les protéines, l'ADN, l'ARN et les ligands avec une précision sans précédent. Cela signifie,AlphaFold 3 accélérera davantage la conception de médicaments et la recherche génomique.Inauguration d'une nouvelle ère de biologie cellulaire basée sur l'intelligence artificielle.
Avec la sortie d'AlphaFold 3,La série Alphafold a finalement construit une base entièrement atomique.De même, la série RoseTTAFold a également lancé avec succès RoseTTAFold All-Atom au cours du premier semestre de cette année, réalisant la capacité de faire des prédictions raisonnables sur les modifications covalentes des protéines et l'assemblage de plusieurs chaînes d'acides nucléiques et de petites molécules.
Avec l’aide d’Alphafold3 et de RoseTTAFold All-Atom, les chercheurs exploitent pleinement le pouvoir de l’imagination. Par exemple, en juin 2024, une équipe de recherche internationale a publié un article dans Nature Biotechnology, démontrant comment utiliser la stratégie de combinaison d'AlphaFold 3 et de RoseTTAFold All-Atom pour concevoir avec succès un nouveau type d'échafaudage protéique capable de délivrer plus efficacement des médicaments directement aux cellules malades, améliorant ainsi les effets thérapeutiques et réduisant les effets secondaires. Cette découverte marque une avancée majeure dans l’application de l’IA en médecine de précision.
Malheureusement, en août 2023, Meta a dissous l'équipe ESMFold et a consacré toute son attention à la promotion de la commercialisation de l'IA. Mais les recherches sur la série ESM ne se sont pas arrêtées. Par exemple, le modèle a réalisé des progrès importants dans le domaine de la modélisation du langage des protéines et a fourni une solution de modélisation unifiée qui intègre des informations multi-échelles. Il s’agit notamment du premier modèle de langage pré-entraîné à base de protéines capable de traiter à la fois les informations sur les acides aminés et les informations atomiques.
On peut voir à partir de là queDans la nouvelle ère où les séries Alphafold, RoseTTAFold et ESM sont à égalité,La combinaison de l’IA et de la recherche sur les protéines va se rapprocher, ce qui non seulement accélérera notre compréhension de la structure et de la fonction des protéines, mais apportera également des changements révolutionnaires au traitement des maladies, au développement de médicaments et aux applications biotechnologiques. Avec le développement rapide apporté par la technologie de l’IA, le domaine de la biologie structurale devient plus dynamique et un nouveau chapitre dans le domaine de la biomédecine s’ouvre lentement.








