Command Palette
Search for a command to run...
Sélectionné Pour l'ICML ! L'équipe De l'Université Renmin a Utilisé Le Réseau Neuronal À Graphe Équivariant Pour Prédire Les Sites De Liaison Des Protéines Cibles, Avec La Plus Grande Amélioration Des Performances De 20%
Dans les systèmes vivants, presque tous les processus biologiques et pharmacologiques impliquent des interactions entre des récepteurs (protéines cibles) et des ligands (petites molécules). Ces interactions se produisent dans des régions spécifiques de la structure de la protéine cible.Connus sous le nom de « sites de liaison », la prédiction des sites de liaison des protéines cibles joue un rôle fondamental dans diverses tâches en aval telles que la découverte de médicaments.
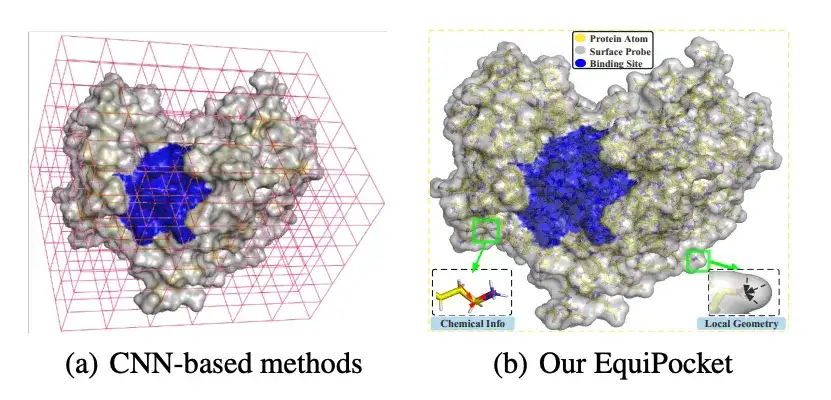
Ces dernières années, inspirés par les avancées de l’apprentissage profond, les réseaux de neurones convolutifs (CNN) ont été appliqués avec succès à la prédiction des sites de liaison des ligands. Les méthodes basées sur CNN traitent les protéines comme des images 3D en regroupant spatialement les atomes de protéines dans les voxels les plus proches, puis modélisent la prédiction du site de liaison comme un problème de détection d'objet ou une tâche de segmentation sémantique sur une grille 3D. Ces méthodes présentent certains avantages, mais il reste encore des défis à relever, tels que :Il présente des défauts dans la représentation des structures protéiques irrégulières ; il est sensible à la rotation ; il ne décrit pas de manière adéquate les caractéristiques géométriques des surfaces protéiques ; et il est insensible aux changements de taille des protéines.
À cette fin, une équipe de recherche de la Gaoling School of Artificial Intelligence de l'Université Renmin de Chine a récemment publié un article de recherche intitulé « EquiPocket : un réseau neuronal à graphes géométriques équivariants E(3) pour la prédiction du site de liaison du ligand » à l'ICML 2024, la principale conférence universitaire dans le domaine de l'IA. Cette étude est la première à appliquer le réseau neuronal à graphe isovariant E(3) (GNN) à la prédiction du site de liaison du ligand.A proposé un cadre appelé EquiPocket,Les défis rencontrés par les méthodes basées sur CNN sont abordés.
Points saillants de la recherche :
* La première application du GNN isovariant E(3) à la prédiction du site de liaison du ligand
* Comparé aux méthodes traditionnelles basées sur CNN, EquiPocket ne nécessite pas de voxélisation, peut modéliser des structures protéiques irrégulières et est insensible à toute transformation euclidienne, résolvant ainsi des problèmes tels que les « défauts de représentation des structures protéiques irrégulières » et la « sensibilité à la rotation »
* Des expériences approfondies sur des méthodes de référence représentatives démontrent la supériorité d'EquiPocket sur les méthodes de pointe actuelles, ce qui peut être utile pour diverses tâches en aval telles que la découverte de médicaments
Adresse du document :
https://openreview.net/forum?id=1vGN3CSxVs
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : Vérification complète de plusieurs ensembles de données professionnelles
Les chercheurs ont sélectionné plusieurs ensembles de données spécialisés et les ont évalués à l’aide d’un sous-ensemble de mlig de chaque ensemble de données contenant des ligands pertinents pour la prédiction du site de liaison.
dans,scPDB est un ensemble de données bien connu pour la prédiction des sites de liaison.Contient des protéines, des ligands et des structures de cavité 3D générées par VolSite. Cette étude a utilisé la version 2017 pour la formation et la validation croisée, qui contient 17 594 structures, 16 034 entrées, 4 782 protéines et 6 326 ligands.
PDBbind est un ensemble de données couramment utilisé pour étudier les complexes protéine-ligand.Contient la structure 3D de la protéine, du ligand, du site de liaison et des résultats d'affinité de liaison précis déterminés en laboratoire. Cette étude a utilisé la version 2020 pour l’évaluation, qui se compose de deux parties : un ensemble commun (14 127 complexes) et un ensemble raffiné (5 316 complexes). L'ensemble général contient tous les complexes protéine-ligand, et l'ensemble raffiné sélectionne les composés de meilleure qualité de l'ensemble général pour les tests expérimentaux.
COACH 420 et HOLO4K sont deux ensembles de données de test utilisés pour la prédiction du site de liaison.Introduit pour la première fois par (Krivák & Hoksza, 2018).
Architecture du modèle : le cadre global d'EquiPocket se compose de trois modules
Le cadre global d'EquiPocket se compose de 3 modules :Comme le montre la figure suivante :
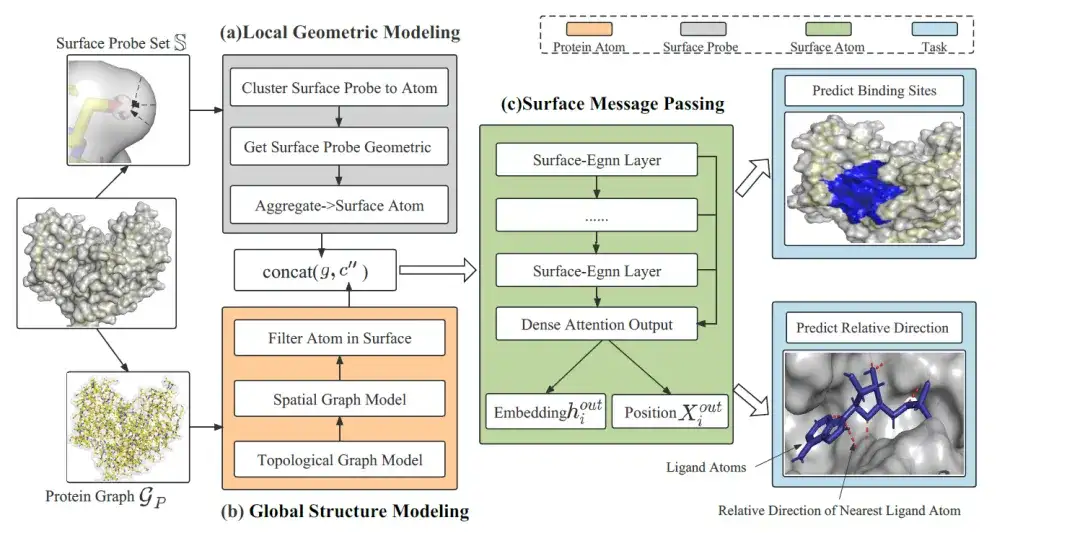
Le premier module est le module de modélisation géométrique locale, qui permet d'extraire les informations géométriques locales de chaque atome de surface ; le deuxième module est le module de modélisation de la structure globale, qui est utilisé pour décrire la structure chimique et spatiale des protéines ; le dernier module est le module Surface Message Passing, qui capture la géométrie de surface en transmettant des informations équivariantes sur les atomes de surface.
Module de modélisation géométrique locale
La géométrie locale de chaque atome de protéine détermine si sa région proche est apte à faire partie du site de liaison.
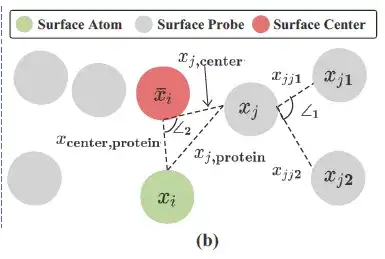
Comme le montre la figure ci-dessus, les chercheurs ont utilisé des sondes de surface (en gris sur la figure ci-dessus) autour de chaque atome de surface de protéine (atome de surface, vert sur la figure ci-dessus) pour décrire les informations géométriques locales. Plus précisément, pour chaque atome de surface i ∈ VS, les sondes de surface qui l'entourent sont renvoyées par un sous-ensemble de S, à savoir :
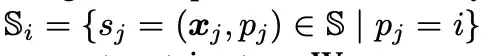
Les chercheurs ont construit des informations géométriques basées sur Si et ont enregistré la valeur centrale/moyenne de toutes les coordonnées 3D dans Si (Surface Centere, rouge dans la figure ci-dessus) comme xi¯.
Module de modélisation structurelle globale
Bien que les sites de liaison soient principalement composés d'atomes de surface, la structure globale de la protéine affecte souvent les interactions des ligands ainsi que la formation du site de liaison et doit donc être modélisée.
Les chercheurs ont atteint cet objectif grâce à deux processus connectés : la modélisation de graphes chimiques et la modélisation de graphes spatiaux. Le module de modélisation structurelle globale résultant est responsable du traitement des informations sur l'ensemble de la protéine, y compris les types d'atomes, les liaisons chimiques, les positions spatiales relatives, etc.
Module de transfert d'informations de surface
Compte tenu des caractéristiques géométriques locales des atomes de surface et des caractéristiques de codage globales, ce module effectuera un transfert d'informations équivariantes sur la carte de surface pour mettre à jour toutes les caractéristiques des atomes de surface des protéines.
Résultats de recherche : EquiPocket améliore les performances de 10-20% par rapport aux modèles de référence
Dans l’expérience, les chercheurs ont choisi de comparer EquiPocket avec les modèles de base suivants :
* Méthodes basées sur la géométrie : Fpocket
* Méthode d'apprentissage automatique : P2rank
* Méthodes basées sur CNN : DeepSite, Kalasanty, DeepSurf, RecurPocket
* Modèles basés sur la topologie : GAT, GCN et GCN2
* Modèles basés sur des graphes spatiaux : SchNet, EGNN
Les mesures utilisées pour évaluer le modèle incluent le DCC (la distance entre le centre du site de liaison prédit et le véritable centre du site de liaison), le DCA (la distance la plus courte entre le centre du site de liaison prédit et toute grille de ligand) et le taux d'échec (le taux d'échantillonnage sans aucun centre de site de liaison prédit). Le tableau ci-dessous montre les résultats de la prédiction du site de liaison sur COACH 420, HOLO4K et PDBbind.
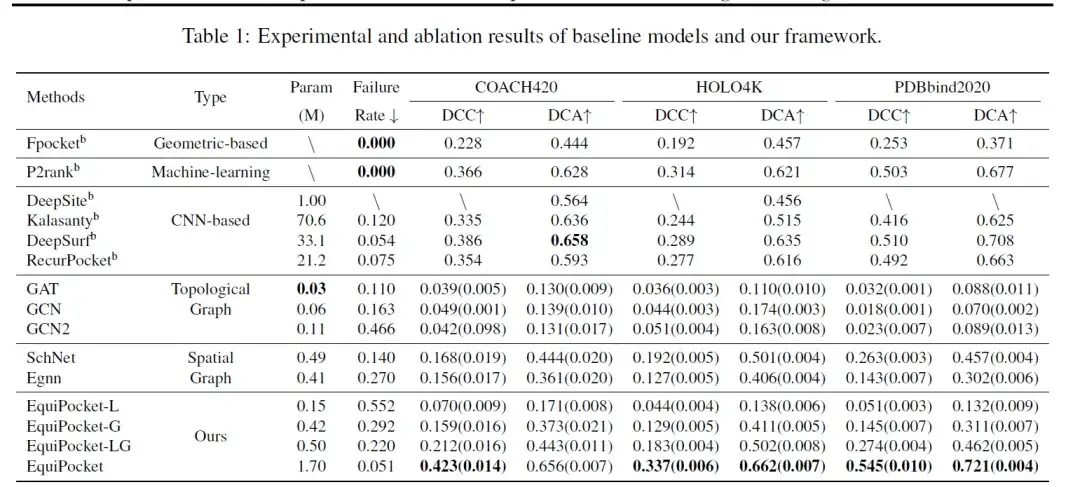
Comme le montrent les données du tableau,La méthode Fpocket basée sur la géométrie a de mauvaises performances.Étant donné que cette méthode utilise uniquement les caractéristiques géométriques de la protéine, le taux d’échec est de 0 ; La méthode d'apprentissage automatique P2rank combine des forêts aléatoires avec les informations géométriques de la surface des protéines, améliorant ainsi considérablement les performances.
Les méthodes basées sur CNN (DeepSite, Kalasanty, DeepSurf, RecurPocket) sont bien plus performantes que les méthodes basées sur la géométrie.Parmi eux, DCC et DCA améliorent plus que 50%, mais nécessitent un grand nombre de paramètres et de ressources de calcul. Parmi elles, les méthodes précédemment proposées, DeepSite et Kalasanty, sont limitées par la variation de la taille des protéines et leur capacité insuffisante à gérer les grosses protéines, ce qui peut conduire à un échec de prédiction.
Pour les modèles graphiques,Les performances des modèles de graphes topologiques (GCN, GAT, GCN2) sont médiocres.C’est principalement parce qu’ils ne prennent en compte que les informations sur les liaisons atomiques et chimiques et ignorent la structure spatiale des protéines ; les performances des modèles de graphes spatiaux (SchNet, EGNN) sont généralement meilleures que celles des modèles de graphes topologiques. EGNN utilise les propriétés des atomes ainsi que leurs positions spatiales relatives/absolues, ce qui fonctionne mieux ; SchNet met à jour les intégrations en se basant uniquement sur les distances relatives des atomes, mais les performances des modèles de graphes spatiaux sont pires que celles des méthodes basées sur CNN et sur la géométrie, car les premières ne peuvent pas obtenir suffisamment de caractéristiques géométriques et ne peuvent pas résoudre le problème des changements de taille des protéines.
Les résultats ci-dessus montrent queLes informations géométriques et les informations structurelles à plusieurs niveaux de la surface des protéines sont cruciales pour la prédiction du site de liaison.
De plus, cela reflète également les limites du modèle GNN actuel, c'est-à-dire qu'il est difficile de collecter suffisamment d'informations géométriques à partir de la surface des protéines ou que les ressources informatiques requises sont trop importantes, ce qui rend difficile son application aux systèmes macromoléculaires tels que les protéines. Par conséquent, le cadre EquiPocket peut non seulement mettre à jour les informations chimiques et spatiales au niveau atomique, mais également collecter efficacement des informations géométriques sans nécessiter de ressources de calcul excessives.Ses performances s'améliorent par rapport aux résultats précédents de 10-20%.
Des ligands de petites molécules aux macromolécules biologiques, l'IA interprète en profondeur la structure des protéines
Il existe des milliards de machines moléculaires à l'intérieur de chaque cellule végétale, animale et humaine, constituées de molécules telles que des protéines, des acides nucléiques, des sucres, et aucune partie d'entre elles ne peut fonctionner seule - ce n'est qu'en comprenant comment elles interagissent dans des millions de combinaisons que nous pouvons acquérir une compréhension plus profonde de la vie.
En mai de cette année, Google DeepMind a publié le modèle AlphaFold3, capable d'effectuer des prédictions structurelles conjointes pour des complexes comprenant des protéines, des acides nucléiques, des petites molécules, des ions et des résidus modifiés. L’interaction entre les protéines et les ligands de petites molécules est au cœur du mécanisme d’action du médicament. Grâce à son algorithme d’apprentissage profond avancé, AlphaFold3 peut prédire avec précision la structure tridimensionnelle de la liaison protéine-ligand, avec une précision dépassant de loin celle des outils d’amarrage existants.
En termes de développement de nouveaux médicaments,Grâce aux structures protéine-ligand prédites par AlphaFold3, les chercheurs peuvent sélectionner et concevoir plus efficacement de nouveaux candidats médicaments et accélérer le processus de découverte de médicaments. En termes d'optimisation des médicaments existants, cet outil peut également être utilisé pour optimiser les médicaments existants en améliorant leur mode de liaison avec la protéine cible pour améliorer l'efficacité ou réduire les effets secondaires.
En plus des ligands à petites molécules,Les protéines doivent également se combiner avec des macromolécules biologiques telles que l’ADN et les sucres pour exercer leurs fonctions biologiques.Actuellement, des milliers de complexes de structures protéiques ont été déposés dans la base de données des protéines grâce à des méthodes expérimentales. Cependant, les méthodes expérimentales traditionnelles sont longues et coûteuses, tandis que les méthodes de prédiction basées sur l’apprentissage automatique peuvent facilement résoudre les défis.
En février de cette année, une équipe de recherche de l'Université agricole de Nanjing a publié un article de recherche en ligne intitulé « ULDNA : intégration de modèles de langage multi-sources non supervisés avec le réseau d'attention LSTM pour une prédiction de site de liaison protéine-ADN de haute précision » dans Briefings in Bioinformatics, une revue importante dans le domaine de la biologie.Une nouvelle méthode de prédiction d'apprentissage profond ULDNA a été développée pour prédire les sites de liaison protéine-ADN.
Adresse du document :
https://academic.oup.com/bib/article/25/2/bbae040/7606634

L'idée principale d'ULDNA est d'utiliser le modèle de langage protéique pour concevoir une représentation des caractéristiques de la séquence, puis de combiner le réseau de mémoire à long terme (LSTM-Attention Network) avec le mécanisme d'attention pour former le modèle de prédiction du site de liaison à l'ADN. Les chercheurs ont sélectionné sept ensembles de données de référence, dont PDNA-128, PDNA-316 et PDNA-335 (avec un nombre de séquences protéiques allant de 40 à 600), et ont effectué un test complet sur ULDNA. Les résultats expérimentaux montrent queULDNA fonctionne bien sur tous les ensembles de données et ses performances de prédiction sont nettement meilleures que celles des neuf autres méthodes courantes.
En plus de l'ADN, les sucres sont omniprésents sur les surfaces cellulaires de tous les organismes, où ils interagissent avec de multiples familles de protéines telles que les lectines, les anticorps, les enzymes et les transporteurs pour réguler les processus biologiques clés tels que les réponses immunitaires, la différenciation cellulaire et le développement neuronal.La compréhension du mécanisme d’interaction entre les glucides et les protéines est la base du développement de médicaments à base de glucides.Cependant, la diversité et la complexité des structures glucidiques, en particulier la variabilité de leurs sites de liaison aux protéines, posent des défis à l’acquisition de données expérimentales et à la conception de médicaments.
Tout récemment, une équipe de l’Académie chinoise des sciences a développé un modèle d’apprentissage profond, DeepGlycanSite, qui peut prédire avec précision les sites de liaison du sucre sur une structure protéique donnée. DeepGlycanSite intègre les caractéristiques géométriques et évolutives des protéines dans un réseau neuronal à graphe équivariant profond basé sur l'architecture Transformer.Ses performances dépassent considérablement les méthodes avancées précédentes et peuvent prédire efficacement les sites de liaison de diverses molécules de sucre.
Associé à des études de mutagenèse, DeepGlycanSite révèle un site de reconnaissance du sucre guanosine-5'-diphosphate pour d'importants récepteurs couplés aux protéines G. Ces résultats suggèrent que DeepGlycanSite est utile pour la prédiction du site de liaison du sucre et peut fournir des informations sur les mécanismes moléculaires derrière la régulation des glucides des protéines thérapeutiquement importantes.

L'étude, intitulée « Prédiction très précise du site de liaison des glucides avec DeepGlycanSite », a été publiée dans Nature Communications le 17 juin 2024.
Adresse du document :
https://www.nature.com/articles/s41467-024-49516-2
En résumé, la protéine est une molécule importante dans les organismes vivants et joue un rôle clé dans la structure et la fonction des cellules. L’étude de la structure des protéines est d’une grande importance pour comprendre les processus vitaux, révéler les mécanismes des maladies et développer des médicaments. Aujourd’hui, l’apprentissage automatique ouvre une nouvelle porte aux scientifiques pour comprendre les mystères de la vie.
Références :
1.https://openreview.net/forum?id=1vGN3CSxVs
2.https://mp.weixin.qq.com/s/aGzcr0ncQA-jBy-vTGC35Q
3.https://www.jiqizhixin.com/articles/2024-05-09
4.https://news.njau.edu.cn/2024/0








