Command Palette
Search for a command to run...
Publié Dans La Revue Cell ! Le Groupe De Recherche Dirigé Par Zhang Qiangfeng De l'Université Tsinghua a Développé l'algorithme SPACE, Qui Possède La Principale Capacité De Découverte De Modules Organisationnels Parmi Des Outils Similaires
Bien que les cellules des organismes multicellulaires partagent le même génome, elles présentent une diversité significative en termes de morphologie, d'expression génétique et de fonction en raison de différences dans leurs réseaux internes de régulation génétique et d'échanges de signaux externes avec les cellules voisines du microenvironnement environnant. Afin d'associer les informations sur le type de cellule à sa localisation spatiale dans les tissus, la technologie de transcriptomique spatiale (ST) a vu le jour.Cette technologie permet non seulement d’obtenir des données de transcriptome à haute résolution, mais également de correspondre à des informations de position pour déterminer la distribution spatiale et la relation de position de différents sous-types de cellules ou états transcriptionnels. Il joue un rôle clé dans la compréhension de la structure de la vie, du développement individuel, de l’évolution de la vie et de la définition des maladies.
Ces dernières années, grâce au développement continu de la technologie de transcriptomique spatiale, les chercheurs sont en mesure d’obtenir le profil d’expression génétique des cellules à une résolution unicellulaire tout en conservant les informations de localisation spatiale des cellules dans les tissus. La manière d’utiliser efficacement ces informations spatiales pour identifier les sous-types de cellules spatiales et découvrir les modules tissulaires est devenue une tâche essentielle dans l’analyse des données du transcriptome spatial.
Actuellement, l’analyse des données du transcriptome spatial est confrontée aux deux défis suivants : premièrement, pour l’identification des types de cellules spatiales, de nombreuses études utilisent uniquement les profils d’expression des gènes cellulaires et ignorent les informations de localisation spatiale des cellules. Les recherches menées ces dernières années ont montré que les types de cellules, que l’on pensait à l’origine homogènes, peuvent être subdivisés en plusieurs sous-types en fonction de leur emplacement dans le tissu. Deuxièmement, pour le type de cheveux des modules tissulaires, étant donné que les caractéristiques d’expression génétique des différentes cellules constituant le tissu peuvent être très hétérogènes, les méthodes d’analyse précédentes n’ont pas réussi à utiliser pleinement l’hétérogénéité des types de cellules dans les données de transcriptome spatial à résolution unicellulaire.
Sur cette base,Groupe de recherche du professeur associé Zhang Qiangfeng à l'École des sciences de la vie, Université Tsinghua/Centre d'innovation avancée en biologie structurale/Centre conjoint des sciences de la vie Tsinghua-Université de Pékin,Un article de recherche intitulé « Découverte de modules tissulaires dans les données de transcriptomique spatiale à résolution unicellulaire via l'intégration cellulaire consciente de l'interaction cellule-cellule » a récemment été publié en ligne dans la revue Cell Systems.
L'étude a développé un algorithme d'intelligence artificielle SPACE (analyse de données transcriptomiques spatiales via l'intégration de cellules « sensibles aux interactions ») basé sur le cadre d'apprentissage profond de l'autoencodeur graphique.La capacité d’identifier les types de cellules spatiales et de découvrir des modules tissulaires à partir de données de transcriptome spatial à une résolution unicellulaire peut être utilisée pour des études de transcriptome spatial à grande échelle.
Points saillants de la recherche :
* Développement de SPACE, un outil d'analyse du transcriptome spatial basé sur l'IA qui peut identifier les types de cellules spatiales et découvrir des modules tissulaires à partir de données de transcriptome spatial à une résolution de cellule unique.
* SPACE surpasse considérablement les autres outils d'identification des types cellulaires et de découverte de modules tissulaires, en particulier dans les tissus complexes contenant plusieurs types cellulaires
* SPACE peut être utilisé pour des études de transcriptome spatial à grande échelle afin de comprendre comment les interactions entre cellules spatialement voisines affectent les fonctions biologiques des types de cellules et des modules tissulaires
Adresse du document :
https://www.cell.com/cell-systems/fulltext/S2405-4712(24)00124-8
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensembles de données : plusieurs ensembles de données vérifient les capacités de SPACE
Afin de vérifier les capacités de SPACE, plusieurs ensembles de données ont été utilisés dans l'étude, qui sont résumés comme suit :
Adresse de téléchargement du jeu de données :
https://go.hyper.ai/CBJfX
Ensemble de données PMC de souris MERFISH
Pour l'ensemble de données PMC de souris MERFISH, la matrice cellule-gène normalisée transformée en logarithme a été obtenue à partir de la bibliothèque d'images cérébrales et les cellules étiquetées comme « autres » ou situées en dehors de la région principale de l'échantillon ont été supprimées.
Lien vers le jeu de données :
Ensemble de données PLA pour souris STARmap
Pour l'ensemble de données PLA de souris STARmap, la matrice cellule-gène normalisée a été fournie par l'article original et transformée en logarithme.
Lien vers le jeu de données :
https://drive.google.com/file/d/1DDCowUuZ7PPFUSZsjvSqntWkYJMjf1Na/view?usp=sharing
Ensemble de données AB sur la souris MERFISH
Pour l'ensemble de données AB de la souris MERFISH, la matrice de comptage des gènes a été obtenue à partir de la bibliothèque CELL x GENE. Le nombre total de cellules par cellule a été normalisé à 10 000 et la matrice cellule-gène normalisée a été transformée logarithmiquement.
Lien vers le jeu de données :
https://cellxgene.cziscience.com/collections/31937775-06024e52-a799-b6acdd2ba2e
Ensemble de données WB de souris MERFISH
Pour l'ensemble de données WB de souris MERFISH, la matrice cellule-gène normalisée transformée en logarithme a été obtenue à partir du référentiel GitHub.
Lien vers le jeu de données :
https://github.com/AllenInstitute/abc_atlas_access
Ensemble de données sur la BC humaine Xenium
Pour l'ensemble de données BC humaines Xenium, la matrice de comptage des gènes a été obtenue à partir du site Web de génomique 10x. Le nombre total de cellules par cellule a été normalisé à 10 000 et la matrice cellule-gène normalisée a été transformée logarithmiquement.
Lien vers le jeu de données :
https://www.10xgenomics.com/products/xenium-in-situ/preview-dataset-human-breast
Ensemble de données CosMx sur le CPNPC humain
Pour l'ensemble de données NSCLC humain CosMx, la matrice cellule-gène normalisée transformée en logarithme a été obtenue à partir du site Web nanoString.
Lien vers le jeu de données :
https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/nsclc-ffpe-dataset
Ensemble de données sur le cerveau humain Visium
Pour l'ensemble de données cérébrales humaines Visium, la matrice de comptage des gènes a été obtenue à l'aide du package Bioconductor spatialLIBD. Les 3 000 gènes hautement variables les plus importants ont été identifiés dans chaque échantillon de l'ensemble de données du cerveau humain Visium à l'aide de la fonction scanpy.pp.highly_variable_genes() (flavor = « seurat_v3 ») du package Python SCANPY (v1.9.1). Le nombre total de cellules par cellule a ensuite été normalisé à 10 000 et la matrice cellule-gène normalisée a été transformée logarithmiquement.
Lien vers le jeu de données :
https://bioconductor.org/packages/release/data/experiment/html/spatialLIBD.html
Architecture du modèle : un modèle intégré aux cellules prenant en compte les interactions cellule-cellule
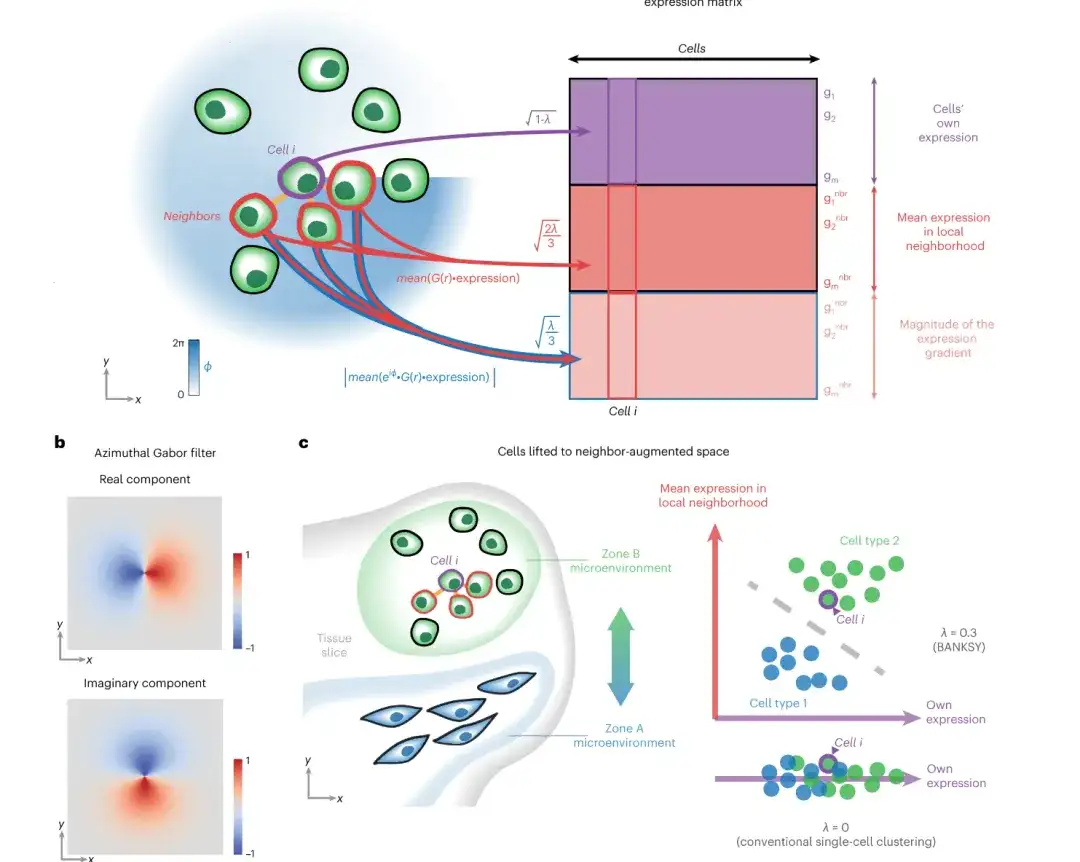
SPACE utilise un cadre d'autoencodeur graphique pour apprendre les intégrations cellulaires de faible dimension qui décrivent les informations d'expression génétique de chaque cellule dans les données du transcriptome spatial ainsi que ses informations d'interaction avec les cellules voisines spatiales (d'où le nom d'intégration cellulaire intégration cellulaire consciente de l'interaction cellule-cellule). Sur la base de cette intégration cellulaire, SPACE utilise ensuite des algorithmes de clustering pour identifier les sous-types de cellules spatiales et découvrir les modules tissulaires.
Du point de vue de l'architecture,Le modèle SPACE se compose de trois parties : un encodeur (réseau d'attention graphique à trois couches), un décodeur de graphe voisin et un décodeur d'expression génétique.La figure suivante montre le cadre général du modèle :
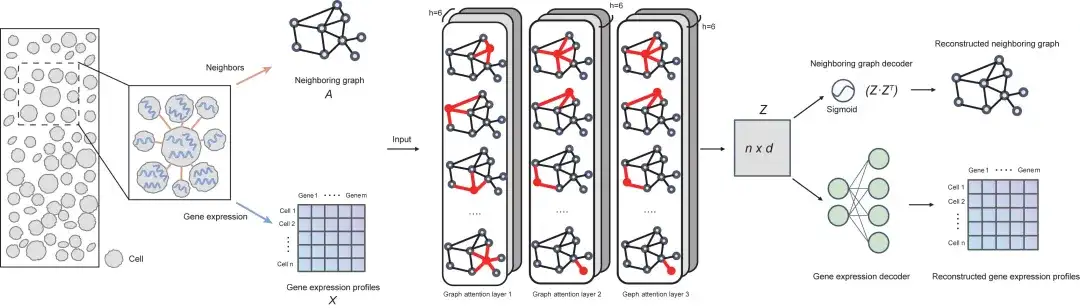
Tout d’abord, SPACE construit un graphe d’adjacence en connectant chaque cellule à ses k cellules voisines les plus proches en fonction de la proximité spatiale ; ensuite, SPACE utilise un réseau d'attention graphique à trois couches (GAT) comme encodeur pour convertir l'entrée du profil d'expression génétique et le graphique d'adjacence en représentations cellulaires de faible dimension, qui sont ensuite utilisées pour reconstruire le profil d'expression génétique et le graphique d'adjacence de chaque cellule via deux réseaux de décodeurs indépendants.
Pour former le modèle GAE, SPACE utilise l'apprentissage auto-supervisé, visant à minimiser la perte totale de reconstruction des profils d'expression génétique et des graphiques d'adjacence. Les représentations cellulaires apprises peuvent ensuite être utilisées pour l’identification du type de cellule et la découverte de modules organisationnels à l’aide de divers algorithmes de clustering.
Les outils d'apprentissage en profondeur développés précédemment ont utilisé des réseaux convolutifs de graphes (GCN) (par exemple, SpaGCN, SpaceFlow, GraphST et SEDR) ou des autoencodeurs d'attention de graphes (par exemple, STAGATE) pour générer des intégrations « sensibles au voisinage » qui découvrent des modules organisationnels en agrégeant les profils d'expression génétique des cellules et de leurs voisins. SPACE diffère de ces outils de trois manières principales :
Tout d’abord, SPACE doit reconstruire à la fois les profils d’expression génétique et les graphiques d’adjacence à partir de la même représentation cellulaire de faible dimension (via deux décodeurs indépendants).Cette conception permet à SPACE de mémoriser les profils d’expression génétique et les interactions spatiales de la cellule analysée et de ses cellules voisines individuelles. En revanche, d’autres méthodes prennent le graphe d’adjacence comme entrée mais ne reconstruisent pas le graphe. Pour souligner cette distinction, cette étude appelle les intégrations cellulaires générées par SPACE « intégrations cellulaires conscientes de l’interaction cellule-cellule ».
Deuxièmement, SPACE définit un rapport de domaine réceptif pour déterminer les poids relatifs du profil d’expression génétique et les pertes de reconstruction du graphique d’adjacence.Ce ratio ajustable permet à SPACE d'adapter l'orientation de l'apprentissage aux besoins de recherche spécifiques, en mettant l'accent sur le profil d'expression génétique de chaque cellule analysée ou sur les interactions des cellules spatialement voisines.
Troisièmement, SPACE utilise également le mécanisme d’attention de l’encodeur GAT pour apprendre de manière adaptative le poids de chaque voisinage pendant le processus d’agrégation des informations de voisinage.Cette approche prend automatiquement en compte les contributions respectives des différents quartiers dans la reconstruction des profils d’expression des gènes.
Résultats de recherche : SPACE surpasse d’autres outils similaires dans l’identification des types de cellules et la découverte de modules tissulaires
SPACE a été testé à l'aide de plusieurs ensembles de données de transcriptome spatial, démontrant que les communautés cellulaires découvertes par SPACE étaient similaires en termes de caractéristiques de distribution spatiale aux structures tissulaires annotées manuellement.
Évaluation de la capacité de SPACE à identifier les types de cellules spatialement informatives
Nous avons initialement utilisé un ensemble de données ST caractérisé par MERFISH du cortex moteur primaire de la souris (PMC) (à partir de la tranche 153) pour étudier la capacité de SPACE à identifier les types de cellules. Les résultats montrent queLes types de cellules identifiés par SPACE correspondent bien à ceux rapportés dans l’étude originale.Comme le montre la figure ci-dessous ; De plus, SPACE fournit également des annotations de type cellulaire à plus haute résolution pour certains types de cellules (tels que les astrocytes et les oligodendrocytes).
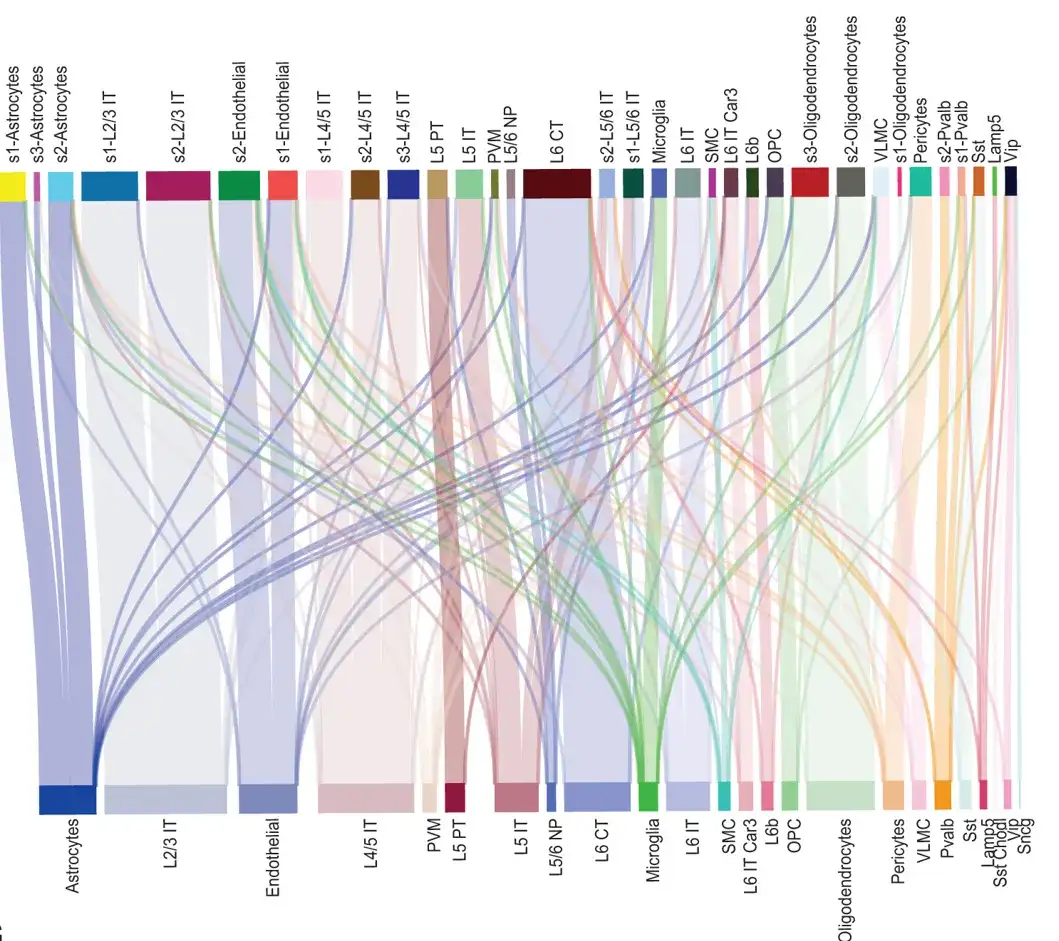
Diagramme de Sankey montrant la correspondance entre les types de cellules pertinents et les types de cellules d'origine dans les informations spatiales de toutes les cellules de la tranche 153 de l'ensemble de données PMC de souris MERFISH
Les chercheurs se sont ensuite concentrés sur les sous-types identifiés d’astrocytes (cellules gliales du cortex) et d’oligodendrocytes (cellules de la gaine de myéline du système nerveux central). Les astrocytes étaient autrefois considérés comme un type de cellule homogène, mais des études récentes sur les astrocytes indiquent qu'ils ont des fonctions distinctes dans différentes régions du cerveau.
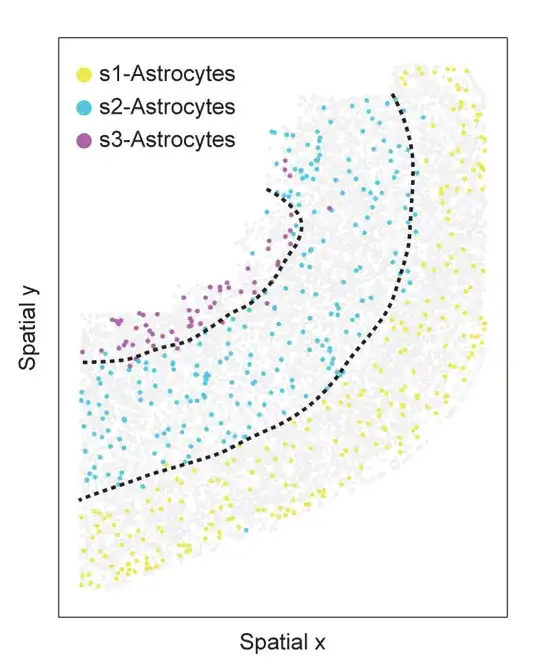
Sous-types d'astrocytes liés aux informations spatiales dans la tranche 153 de l'ensemble de données PMC de souris MERFISH. Les cellules sont colorées par sous-type d’astrocytes et les points gris clair représentent les autres cellules. Les lignes pointillées indiquent la matière supérieure, profonde et blanche
Dans l'expérience, SPACE a trouvé trois sous-types différents dans la tranche PMC 153, comme le montre la figure ci-dessus, chaque sous-type était distribué spatialement dans une couche corticale différente. Semblable aux astrocytes, SPACE classe également les oligodendrocytes en trois sous-types spatialement informatifs avec des modèles de distribution spatiale distincts.

Les chercheurs ont également appliqué SPACE à un ensemble de données de placenta de souris (PLA) généré par STARmap, une autre technologie ST. Les résultats montrent que SPACE a étiqueté les cellules en 16 types de cellules, qui correspondent bien aux types de cellules de l’étude originale, comme indiqué ci-dessus. SPACE a identifié deux sous-types de glycotrophoblastes, tous deux annotés comme cellules « mégatrophoblastes 2 » dans l’étude originale. Les deux sous-types sont localisés dans des régions distinctes du placenta et possèdent des types de cellules voisines uniques en interaction.
En résumé, l’analyse de deux ensembles de données indépendants basés sur différentes méthodes et organisations ST soutient les conclusions suivantes :SPACE est capable d'identifier des types de cellules biologiquement distincts sur la base des informations spatiales contenues dans l'ensemble de données ST.
Évaluation des performances de SPACE dans l'identification des types cellulaires
Les chercheurs ont comparé SPACE avec deux outils actuels d’identification des types de cellules à partir de données transcriptomiques spatiales, BANKSY et FICT, qui prennent en compte les informations spatiales en plus de l’expression des gènes. Dans leur analyse, les chercheurs ont également inclus SCANPY, un outil largement utilisé pour l’identification des types de cellules, bien qu’il ne prenne en compte que l’expression des gènes.
À titre de comparaison, les chercheurs ont utilisé l’ensemble de données PMC de souris MERFISH mentionné précédemment et l’ensemble de données PLA de souris STARmap. Comme le montre la figure ci-dessous, SPACE est capable d'identifier différents sous-types d'astrocytes et d'oligodendrocytes informés spatialement, mais ni SCANPY ni FICT ne peuvent définir des sous-types d'astrocytes et d'oligodendrocytes avec des modèles de distribution spatiale résolus par la couche corticale.
Pour l'ensemble de données PLA de souris STARmap, bien que SPACE et BANKSY aient identifié avec succès les deux sous-types de glycotrophoblastes, SCANPY et FICT n'ont pas réussi à identifier les sous-types de glycotrophoblastes, ce qui peut être dû aux différences évidentes dans les types de cellules environnantes entre les deux sous-types de glycotrophoblastes.
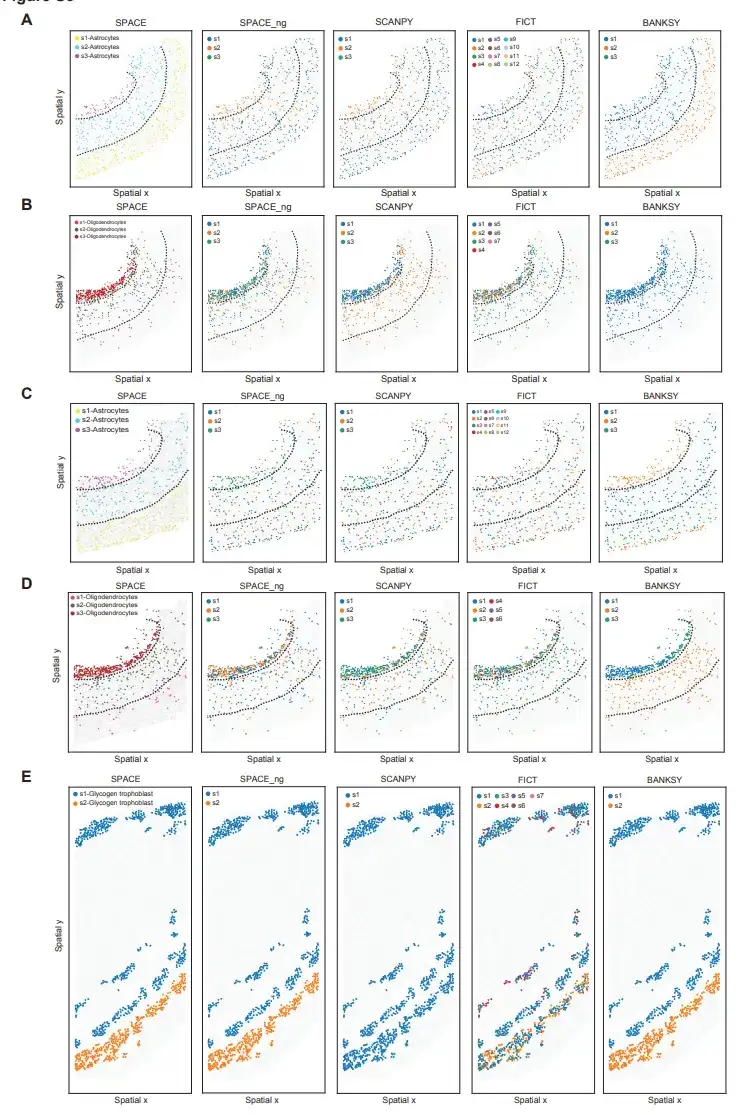
Ces résultats indiquent collectivement queSPACE surpasse les outils actuellement disponibles pour distinguer les types de cellules spatialement informatives des données ST.
SPACE surpasse les outils de pointe dans la découverte de modules tissulaires
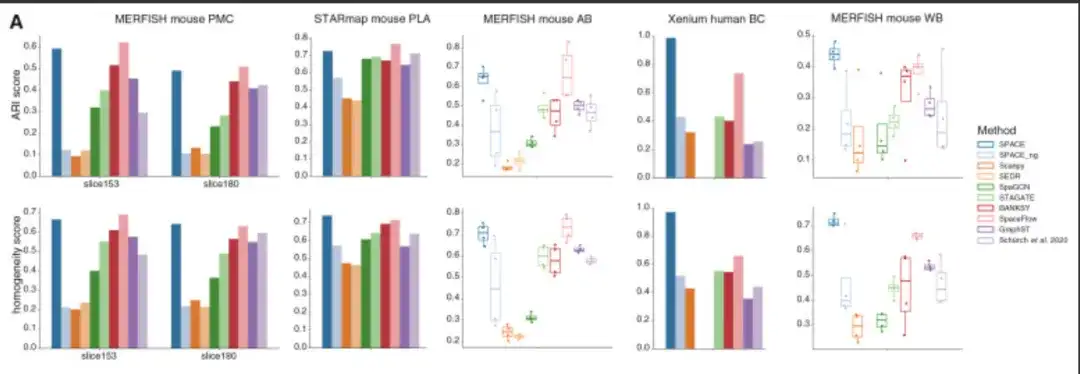
Une tâche importante dans les études de transcriptomique spatiale est de découvrir des modules d’organisation au sein d’un tissu donné. Pour évaluer les capacités de SPACE à cet égard, les chercheurs ont comparé SPACE avec SEDR, SpaGCN, STAGATE, BANKSY, SpaceFlow, GraphST, les méthodes de Schürch et al., ainsi que SCANPY et SPACE_ng, et ont utilisé deux des ensembles de données ST susmentionnés (ensemble de données PMC de souris MERFISH et ensemble de données PLA de souris STARmap), ainsi que trois ensembles de données supplémentaires avec des modules de tissus annotés, y compris l'ensemble de données du cerveau âgé de souris MERFISH (AB), l'ensemble de données du cerveau entier de souris MERFISH (WB) et l'ensemble de données du cancer du sein humain Xenium (BC), qui représentent des données ST obtenues à partir de différents tissus et dans différentes conditions.
Dans l'ensemble,SPACE surpasse largement les outils concurrents dans 2 des 5 ensembles de données et fonctionne presque aussi bien que les outils les plus performants (par rapport aux meilleurs outils respectifs) dans les 3 autres ensembles de données.Comme le montre la figure suivante :
Surmonter les défis de l'analyse des données du transcriptome spatial
La technologie de transcriptomique spatiale est l’une des avancées majeures dans le domaine de la bioinformatique ces dernières années et a été nommée Technologie de l’année par Nature Method en 2020.Cette technologie compense le défaut de la technologie de séquençage de cellules individuelles qui a du mal à mesurer la relation de position entre les cellules individuelles en mesurant simultanément la position spatiale d'un grand nombre de cellules et le nombre de transcriptomes dans les cellules, fournissant ainsi une nouvelle base de données pour comprendre les interactions entre plusieurs cellules. Le développement de méthodes d’analyse de base pour les données de transcriptome spatial est l’un des problèmes de pointe actuels dans le domaine de la bioinformatique.
Le couplage des informations de localisation spatiale cellulaire et de leur spectre caractéristique moléculaire a produit un nouveau type de ressource de données multimodale à haut débit, qui pose de nombreux défis au développement de méthodes efficaces d'analyse de données et d'exploration d'informations. L’intelligence artificielle apporte de nouvelles idées pour résoudre ces défis.

En juillet 2022, le groupe de recherche du professeur Shen Hongbin et du professeur associé Yuan Ye du département d'automatisation de l'école d'information électronique et de génie électrique de l'université Jiao Tong de Shanghai a publié un article de recherche intitulé « Regroupement cellulaire pour les données transcriptomiques spatiales avec des réseaux de neurones graphiques » dans Nature Computational Science, une filiale de Nature.
Lien vers l'article :https://www.nature.com/articles/s43588-022-00266-5
L'article propose une méthode de clustering cellulaire du transcriptome spatial (Cell Clustering for Spatial Transcriptomics, CCST) basée sur un réseau neuronal convolutif graphique.Il fournit une nouvelle solution pour le traitement des données de transcriptome spatial et a le potentiel d'être appliqué à la recherche de problèmes fondamentaux à plusieurs niveaux dans les sciences de la vie et médicales, y compris la modélisation de la distribution spatiale de l'expression des gènes, l'analyse de la dynamique cellulaire et la découverte des interactions clés entre les sous-types cellulaires et leurs mécanismes moléculaires.
Avril 2023Une équipe de recherche de l’Université Johns Hopkins a développé SpaceMarkers.Il s’agit d’un algorithme bioinformatique qui peut utiliser l’analyse de l’espace latent des données ST pour déduire les changements moléculaires dans les interactions cellule-cellule. Les chercheurs ont utilisé cette approche pour déduire les changements moléculaires dans les interactions tumeur-système immunitaire dans les lésions métastatiques, invasives et précurseurs, ainsi qu'en réponse à l'immunothérapie en utilisant les données transcriptomiques spatiales de Visium.
L'étude a été publiée dans Cell Systems sous le titre « Découvrir le paysage spatial des interactions moléculaires au sein du microenvironnement tumoral à travers des espaces latents ».
En avril de cette année, un rapport de recherche intitulé « BANKSY unifie le typage cellulaire et la segmentation des domaines tissulaires pour une analyse de données omiques spatiales évolutive » a été publié dans la revue internationale Nature Genetics.Des scientifiques de l'A*STAR Research Institute de Singapour et d'autres institutions ont présenté un algorithme appelé BBANKSY (Building Aggregates with a Neighborhood Kernel and Spatial Yardstick).En tant qu'outil innovant d'analyse de données omiques spatiales, la fonction principale de cet algorithme est de classer efficacement les cellules dans les données omiques spatiales en fonction du type et du domaine tissulaire.
Lien vers l'article :https://www.nature.com/articles/s41588-024-01664-3
De toute évidence, avec le soutien de la technologie de l'intelligence artificielle à l'avenir, la technologie de transcriptomique spatiale révélera mieux la distribution spatiale de divers types de cellules dans les tissus, les interactions entre diverses populations cellulaires et dessinera des cartes d'expression génétique de différentes régions tissulaires, ce qui a une valeur d'application de grande envergure pour comprendre le mécanisme d'apparition des maladies et des cancers.
Références :
1.https://www.cell.com/cell-systems/fulltext/S2405-4712(24)00124-8#secsectitle0030
2.https://www.tsinghua.edu.cn/info/1175/112190.htm
3.https://news.bioon.com/article/367a820e60b9.html
4.https://www.sohu.com/a/677912398_12








