Command Palette
Search for a command to run...
Sélectionné Pour l'ICML, Tsinghua AIR Et d'autres Ont Publié Conjointement Le Modèle De Langage Protéique ESM-AA, Surpassant Le SOTA Traditionnel

En tant que force motrice d’innombrables réactions biochimiques au sein des cellules, les protéines jouent le rôle d’architectes et d’ingénieurs dans le monde microscopique des cellules. Ils ne catalysent pas seulement les activités vitales, mais servent également de composants de base pour la construction et le maintien de la morphologie et de la fonction des organismes. C’est l’interaction et la synergie entre les protéines qui soutiennent le grand plan de la vie.
Cependant, la structure des protéines est complexe et changeante, et les méthodes expérimentales traditionnelles sont longues et laborieuses pour analyser la structure des protéines. Les modèles de langage des protéines (PLM) ont vu le jour. En utilisant la technologie d'apprentissage profond, en analysant une grande quantité de données de séquences protéiques, en apprenant les lois biochimiques et les modèles de coévolution des protéines, il a réalisé des réalisations remarquables dans les domaines de la prédiction de la structure des protéines, de la prédiction de l'adaptabilité et de la conception des protéines, et a grandement favorisé le développement de l'ingénierie des protéines.
Bien que les PLM aient obtenu un grand succès à l’échelle des résidus, ils sont limités dans leur capacité à fournir des informations au niveau atomique. En réponse à cela, Zhou Hao, chercheur associé à l'Institut des industries intelligentes de l'Université Tsinghua, s'est associé à l'Université de Pékin, à l'Université de Nanjing et à l'équipe moléculaire de Shuimu pourUn modèle de langage protéique multi-échelle ESM-AA (ESM All Atom) est proposé.En concevant des mécanismes de formation tels que l’expansion des résidus et le codage de position multi-échelle, la capacité de traiter des informations à l’échelle atomique a été élargie.
Les performances de l'ESM-AA dans des tâches telles que la liaison cible-ligand ont été considérablement améliorées, surpassant les modèles actuels de langage protéique SOTA tels que l'ESM-2, et surpassant également les modèles actuels d'apprentissage de représentation moléculaire SOTA tels que Uni-Mol. Des recherches connexes ont été publiées sous le titre « ESM All-Atom : modèle de langage protéique multi-échelle pour la modélisation moléculaire unifiée ».Publié à l'ICML, la principale conférence sur l'apprentissage automatique.

Adresse du document :
https://icml.cc/virtual/2024/poster/35119
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : Un ensemble de données mixtes de données protéiques et moléculaires a été construit
Dans la tâche de pré-formation,L’étude a utilisé un ensemble de données combinées de protéines et de données moléculaires contenant des informations structurelles telles que les coordonnées atomiques.
Pour l'ensemble de données protéiques, l'étude a utilisé AlphaFold DB, qui contient 8 millions de séquences et de structures protéiques prédites par AlphaFold2 avec une confiance élevée.
Pour l’ensemble de données moléculaires, l’étude a utilisé des données générées par les champs de force moléculaire ETKDG et MMFF, contenant 19 millions de molécules et 209 millions de configurations.
Lors de la formation de l'ESM-AA, les chercheurs ont d'abord mélangé un ensemble de données protéiques Dp et un ensemble de données moléculaires Dm pour former l'ensemble de données final, c'est-à-dire D=Dp∪Dm. Pour les molécules de Dm, puisqu'elles sont composées uniquement d'atomes, leur séquence de conversion de code X̄ est l'ensemble ordonné de tous les atomes Ā sans aucun résidu, c'est-à-dire R̄=∅. Il convient de noter que, puisque les données moléculaires sont utilisées dans la pré-formation, ESM-AA peut accepter à la fois des protéines et des molécules en entrée.
Construction du modèle ESM-AA : pré-entraînement et codage multi-échelles pour obtenir une modélisation moléculaire unifiée
Inspiré par la méthode de commutation de code multilingue, ESM-AA décompresse d'abord de manière aléatoire certains résidus pour générer des séquences de protéines de commutation de code multi-échelle lors de l'exécution de tâches de prédiction et de conception de protéines. Ces séquences sont ensuite entraînées grâce à des codages positionnels multi-échelles soigneusement conçus, et leur efficacité a été démontrée aux échelles résiduelle et atomique.
Lorsqu'il s'agit de tâches moléculaires protéiques, c'est-à-dire de tâches impliquant des protéines et des petites molécules, ESM-AA ne nécessite aucune assistance de modèle supplémentaire et peut utiliser pleinement les capacités du modèle pré-entraîné.
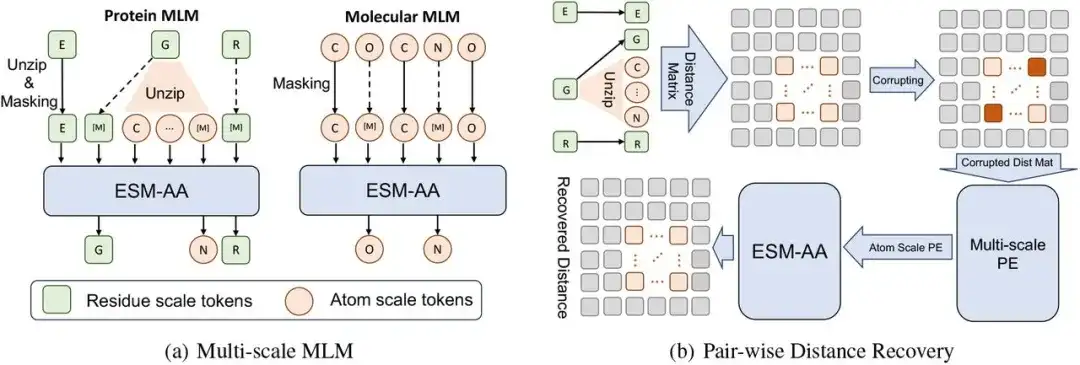
Le cadre de pré-formation multi-échelle de cette étude consiste en un modèle de langage masqué multi-échelle (MLM) et une récupération de distance par paires.
Plus précisément, à l'échelle des résidus, une protéine X peut être considérée comme une séquence de L résidus, c'est-à-dire X = (r1,…,ri,…,rL). Chaque résidu ri est composé de N atomes A Ai={a1i,…,aNi}. Pour construire la séquence protéique de commutation de code X̅, nous avons mis en œuvre un processus de décompression en sélectionnant aléatoirement un ensemble de résidus et en insérant leurs atomes correspondants dans X. Dans ce processus, les chercheurs ont organisé les atomes décompressés dans l'ordre, et finalement, après avoir inséré l'ensemble d'atomes Ai dans X (c'est-à-dire en décompressant le résidu ri), ils ont obtenu une séquence de commutation de code X̄.
Alors,Les chercheurs ont effectué une modélisation du langage masqué sur la séquence de commutation de code X̄.
Tout d’abord, nous masquons aléatoirement certains atomes ou résidus dans X̄ et laissons le modèle prédire les atomes ou résidus d’origine en utilisant le contexte environnant. Les chercheurs ont ensuite utilisé la récupération de distance par paires (PDR) comme autre tâche de pré-formation. Autrement dit, les informations structurelles à l'échelle atomique sont détruites en ajoutant du bruit aux coordonnées, et les informations de distance interatomique détruites sont utilisées comme entrée du modèle, ce qui oblige le modèle à restaurer la distance euclidienne précise entre ces atomes.
Compte tenu de la différence sémantique entre les informations structurelles à longue portée sur différents résidus et les informations structurelles à l'échelle atomique au sein d'un seul résidu, cette étude calcule uniquement le PDR au sein du résidu, ce qui peut également permettre à ESM-AA d'apprendre diverses connaissances structurelles au sein de différents résidus.
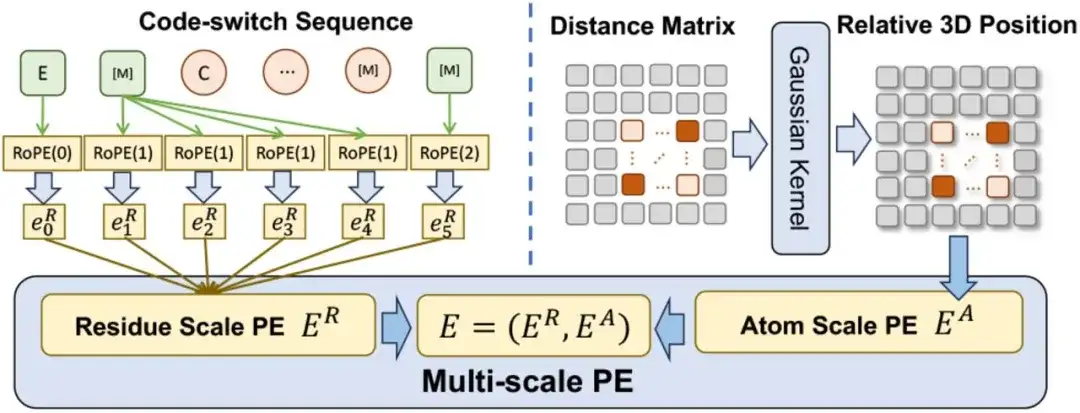
En termes de codage de position multi-échelle, les chercheurs ont conçu un codage de position multi-échelle E pour coder la relation positionnelle dans la séquence de commutation de code. E contient un code de position à l'échelle des résidus ER et un code de position à l'échelle atomique EA.
Pour les urgences,Les chercheurs ont étendu une méthode de codage existante pour lui permettre de coder les relations résidu-atome tout en maintenant la cohérence avec le codage d'origine lorsqu'il s'agit de séquences de résidus purs.Pour EA,Pour capturer les relations entre les atomes, l’étude utilise directement une matrice de distance spatiale pour coder leurs positions tridimensionnelles.
Il convient de mentionner que la méthode de codage multi-échelle garantit que la pré-formation ne sera pas affectée par la relation de position ambiguë, de sorte que l'ESM-AA peut fonctionner efficacement aux deux échelles.
Lors de l'intégration de PE multi-échelle dans Transformer, l'étude a d'abord remplacé le codage sinusoïdal dans Transformer par le codage de position d'échelle résiduelle ER, et a considéré le codage de position d'échelle atomique EA comme le terme de polarisation de la couche d'auto-attention.
Résultats de recherche : Intégrer les connaissances moléculaires pour optimiser la compréhension des protéines
Afin de vérifier l’efficacité du modèle pré-entraîné unifié multi-échelle, cette étude a évalué les performances de l’ESM-AA dans diverses tâches impliquant des protéines et des petites molécules.
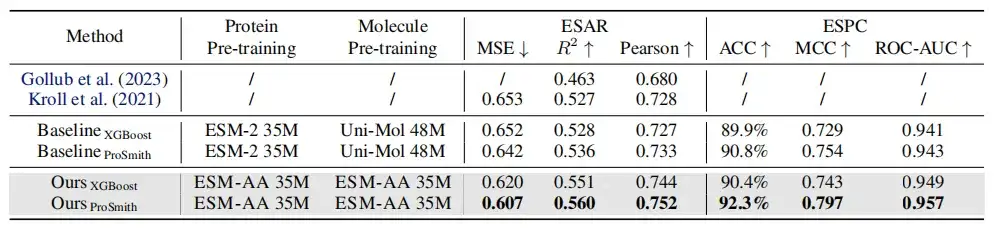
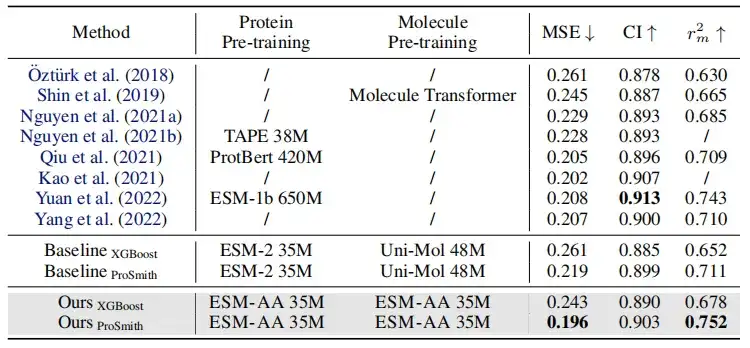
Comme le montre le tableau ci-dessus, dans la comparaison des performances de la tâche de régression d'affinité enzyme-substrat, de la tâche de classification des paires enzyme-substrat et de la tâche de régression d'affinité médicament-cible,Sur la plupart des mesures, ESM-AA surpasse les autres modèles et atteint des résultats de pointe.De plus, les stratégies de réglage fin (telles que ProSmith et XGBoost) basées sur ESM-AA ont systématiquement surpassé les versions combinant deux modèles moléculaires pré-entraînés indépendants avec le modèle protéique pré-entraîné (comme indiqué dans les quatre dernières lignes des tableaux 1 et 2).
Il convient de noter queESM-AA peut même surpasser les méthodes qui utilisent des modèles pré-entraînés avec des tailles de paramètres plus grandes.(Par exemple, la comparaison entre les 5e, 7e et dernière lignes du tableau 2).
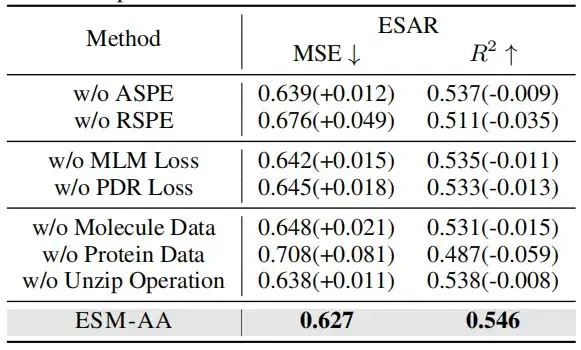
Pour vérifier l'efficacité du codage de position multi-échelle, cette étude a mené des expériences d'ablation dans deux cas : l'un sans utiliser le codage de position à l'échelle atomique (ASPE) ; l'autre est sans utiliser le codage de position d'échelle génétique (RSPE).
Lors de la suppression des données sur les molécules ou les protéines, les performances du modèle ont considérablement diminué. Il est intéressant de noter que la dégradation des performances causée par la suppression des données protéiques est plus évidente que celle causée par la suppression des données moléculaires. Cela suggère que lorsque le modèle n’est pas formé sur des données protéiques, il perd rapidement les connaissances liées aux protéines, ce qui entraîne une baisse significative des performances globales. Cependant,Même sans données moléculaires, le modèle peut toujours obtenir des informations au niveau atomique grâce à des opérations de décompression.
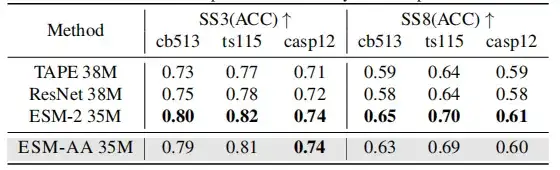
Étant donné que l'ESM-AA a été développé sur la base de PLM existants, cette étude espère déterminer s'il conserve toujours une compréhension complète des protéines, testant ainsi la capacité des modèles pré-entraînés de protéines à comprendre la structure des protéines en utilisant des tâches de prédiction de structure secondaire et de prédiction de contact non supervisée.
Les résultats montrent que même si l’ESM-AA ne peut pas atteindre des performances optimales dans ce type d’étude,Cependant, ses performances en matière de prédiction de structure secondaire et de prédiction de contact sont similaires à celles de l'ESM-2.
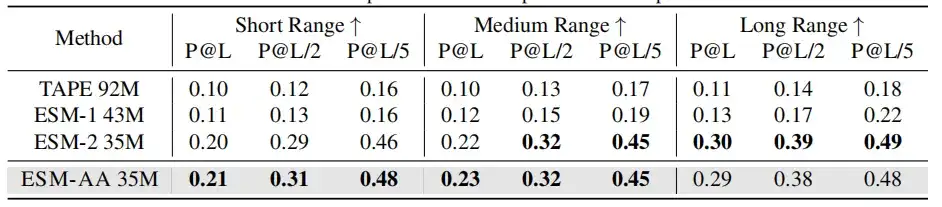
Dans l'analyse comparative moléculaire,L'ESM-AA présente des performances comparables à celles d'Uni-Mol dans la plupart des tâches.Il surpasse plusieurs modèles spécifiques aux molécules dans de nombreux cas, démontrant son potentiel en tant qu'approche puissante pour les tâches moléculaires.
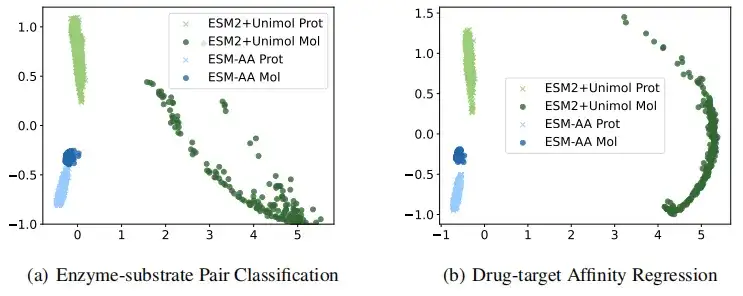
Pour illustrer de manière plus intuitive que l'ESM-AA obtient des représentations de protéines et de petites molécules de meilleure qualité, cette étude a comparé visuellement les représentations extraites par l'ESM-AA et l'ESM-2+Uni-Mol dans les tâches de classification des paires enzyme-substrat et de régression d'affinité cible médicamenteuse. Les résultats montrent queLe modèle ESM-AA est capable de créer une représentation sémantique plus cohérente des données protéiques et moléculaires, ce qui permet à ESM-AA de surpasser les deux modèles pré-entraînés distincts.
Modèle de langage des protéines, la prochaine étape du grand modèle de langage
Depuis les années 1970, de plus en plus de scientifiques pensent que « le XXIe siècle est le siècle de la biologie ». En juillet dernier, Forbes a écrit un long article, imaginant que le LLM place les gens à l’avant-garde d’une nouvelle série de changements dans le domaine de la biologie. La biologie s’avère être un système qui peut être déchiffré, programmé et, dans une certaine mesure, même numérisé.Le LLM, avec son étonnante capacité à contrôler les langues naturelles, offre le potentiel de déchiffrer les langages biologiques.Cela fait également du modèle de langage des protéines l’un des domaines les plus populaires de notre époque.
Les modèles de langage protéique représentent des applications de pointe de la technologie de l’IA en biologie. En apprenant les modèles et les structures des séquences de protéines, il peut prédire la fonction et la morphologie des protéines, ce qui est d’une grande importance pour le développement de nouveaux médicaments, le traitement des maladies et la recherche biologique fondamentale.
Auparavant, les modèles de langage protéique tels que ESM-2 et ESMFold ont démontré une précision comparable à celle d'AlphaFold, avec des vitesses de traitement plus rapides et des capacités de prédiction plus précises pour les « protéines orphelines ». Cela accélère non seulement la prédiction de la structure des protéines, mais fournit également de nouveaux outils pour l’ingénierie des protéines, permettant aux chercheurs de concevoir des séquences de protéines entièrement nouvelles avec des fonctions spécifiques.
De plus, le développement de modèles de langage protéique bénéficie des « lois d’échelle ».Autrement dit, les performances du modèle s’améliorent considérablement avec l’augmentation de l’échelle du modèle, de la taille de l’ensemble de données et de l’effort de calcul.Cela signifie qu’avec l’augmentation des paramètres du modèle et l’accumulation de données de formation, les capacités du modèle de langage protéique réaliseront un saut qualitatif.
Au cours des deux dernières années, les modèles de langage protéique sont également entrés dans une période de développement rapide dans le monde des affaires. En juillet 2023, Baidu Biosciences et l'Université Tsinghua ont proposé conjointement un modèle appelé xTrimo Protein General Language Model (xTrimoPGLM) avec une taille de paramètre allant jusqu'à 100 milliards (100B), qui a considérablement surpassé les autres modèles de base avancés dans plusieurs tâches de compréhension des protéines (13 tâches sur 15).Dans la tâche de génération, xTrimoPGLM est capable de générer de nouvelles séquences de protéines similaires aux structures protéiques naturelles.
En juin 2024, la société de protéines IA Tushen Zhihe a annoncé queLe premier modèle de protéines en langage naturel en Chine, TourSynbio™, a été rendu open source pour tous les chercheurs et développeurs.Ce modèle permet de comprendre la littérature sur les protéines de manière conversationnelle, y compris des fonctions telles que les propriétés des protéines, la prédiction des fonctions et la conception des protéines. En termes d'indicateurs d'évaluation comparant les ensembles de données d'évaluation des protéines, il surpasse GPT4 et devient le premier du secteur.
En outre, la percée dans la recherche technologique représentée par l'ESM-AA peut également signifier que le développement de la technologie est sur le point de dépasser le « moment des frères Wright » et d'inaugurer un bond en avant. Dans le même temps, l’application des modèles de langage protéique ne se limitera pas seulement aux domaines médicaux et biopharmaceutiques, mais pourra également être étendue à de multiples domaines tels que l’agriculture, l’industrie, la science des matériaux et l’assainissement de l’environnement, favorisant l’innovation technologique dans ces domaines et apportant des changements sans précédent à l’humanité.








