Command Palette
Search for a command to run...
Le Modèle De Cellule À 100 Millions De Paramètres Est Ici ! Une Équipe De l'Université Tsinghua Publie scFoundation Dans La Revue Nature : Modélisation Simultanée De 20 000 Gènes
Ces dernières années, les modèles pré-entraînés à grande échelle sont à l’origine d’une nouvelle vague d’intelligence artificielle. Le « grand modèle » extrait des règles de niveau profond à partir de données multi-sources à grande échelle et peut ensuite servir de « modèle de base » pour répondre à une variété de tâches dans différents domaines. Par exemple, les grands modèles linguistiques ont maîtrisé la capacité de comprendre et de reconnaître le langage en apprenant à partir de grandes quantités de données textuelles, révolutionnant ainsi le paradigme dans le domaine du traitement du langage naturel.
De même, dans le domaine des sciences de la vie, les organismes ont également leur propre « langage sous-jacent » : les cellules sont les unités structurelles et fonctionnelles de base du corps humain.Si les valeurs d'expression de l'ADN, de l'ARN, des protéines et des gènes sont comparées à des « mots », elles sont combinées ensemble pour former la phrase « cellule ».Par conséquent, si nous pouvons développer un modèle cellulaire d’intelligence artificielle basé sur le « langage » cellulaire, cela fournira, espérons-le, un nouveau paradigme de recherche et des outils de recherche révolutionnaires pour les sciences de la vie et la médecine.
Cependant,Il existe actuellement trois principaux défis dans la formation de données unicellulaires à grande échelle :
* Les données de pré-formation sur l’expression génétique doivent couvrir des paysages cellulaires de différents états et types. Actuellement, la plupart des données de séquençage d’ARN unicellulaire (scRNA-seq) sont organisées de manière lâche et une base de données complète et exhaustive fait toujours défaut ;
* Pendant l’entraînement, les Transformers traditionnels ont du mal à traiter des « phrases » composées de près de 20 000 gènes codant pour des protéines ;
* Les données scRNA-seq provenant de différentes technologies et de différents laboratoires varient en termes de profondeur de séquençage, ce qui empêche le modèle d'apprendre des représentations cellulaires et génétiques unifiées et significatives.
Pour relever ces défis,Le professeur Zhang Xuegong, directeur du laboratoire des modèles de base de la vie du département d'automatisation de l'université Tsinghua, le professeur Ma Jianzhu du département d'électronique/AIR et le Dr Song Le de biotechnologie ont collaboré à la recherche.En juin 2024, un article de recherche intitulé « Modèle de fondation à grande échelle sur la transcriptomique à cellule unique » a été publié dans Nature Methods.
L’article présente un modèle de grande cellule appelé scFoundation, qui peut traiter environ 20 000 gènes simultanément.En tant que modèle de base, il démontre des améliorations de performances exceptionnelles dans une variété de tâches biomédicales en aval telles que l'amélioration de la profondeur du séquençage cellulaire, la prédiction de la réponse aux médicaments cellulaires et la prédiction des perturbations cellulaires, offrant un nouveau paradigme pour l'intelligence artificielle dans la recherche sur cellule unique.
Points saillants de la recherche :
Le modèle cellulaire scFoundation est formé sur la base de données d'expression génétique provenant de 50 millions de cellules, comporte 100 millions de paramètres et peut traiter environ 20 000 gènes simultanément*. Le modèle utilise une conception asymétrique pour réduire les défis de calcul et de mémoire*. Le modèle fournit de nouvelles idées de recherche pour l'inférence du réseau génétique et l'identification des facteurs de transcription.

Adresse du document :
https://www.nature.com/articles/s41592-024-02305-7
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit également des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensembles de données : création d'un ensemble de données unicellulaire complet
Les chercheurs ont construit un ensemble complet de données unicellulaires en collectant toutes les données de ressources unicellulaires accessibles au public.Il s'agit notamment de Gene Expression Omnibus (GEO), Single Cell Portal, HCA, Human Genome Project (hECA), Deeply Integrated Human Single-Cell Omics Data (DISCO), European Molecular Biology Laboratory-European Bioinformatics Institute Database (EMBL-EBI), etc.
* Adresse de téléchargement GEO :https://www.ncbi.nlm.nih.gov/geo/
* Lien de téléchargement du portail à cellule unique:https://singlecell.broadinstitute.org/single_cell
* Adresse de téléchargement HCA :https://data.humancellatlas.org/
* Adresse de téléchargement EMBL-EBI :https://www.ebi.ac.uk/
Les chercheurs ont aligné toutes les données avec une liste de gènes de 19 264 gènes codant pour des protéines et des gènes mitochondriaux communs identifiés par le Comité de nomenclature des gènes HUGO. Après le contrôle de la qualité des données,Plus de 50 millions de données scRNA-seq humaines ont été obtenues pour la pré-formation.
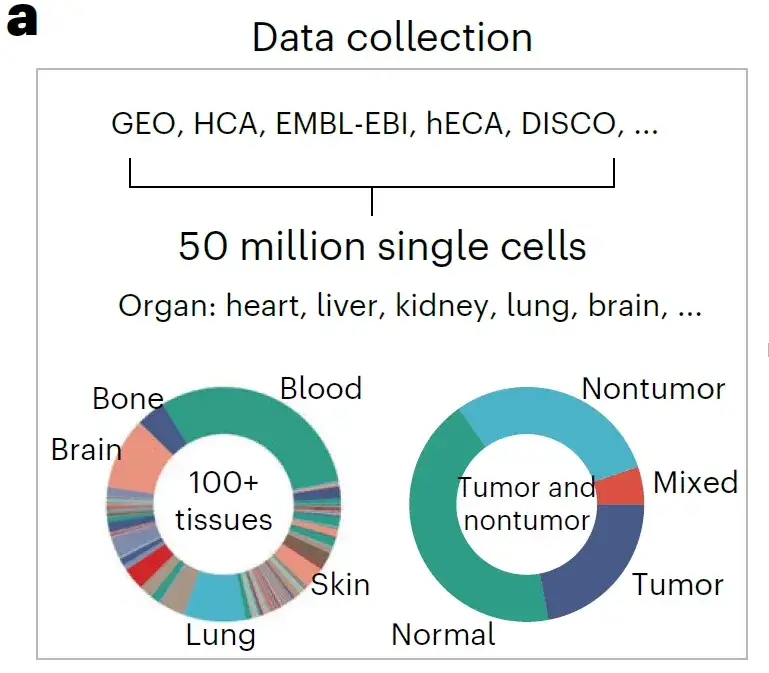
De nombreuses sources de données permettent de construire des ensembles de données de pré-formation riches en modèles biologiques. D'un point de vue anatomique, il couvre plus de 100 types de tissus, couvrant un large éventail de maladies, de tumeurs et d'états normaux, et comme le montre la figure ci-dessus, comprend presque tous les types et états de cellules humaines connus.
Architecture du modèle : création d'un modèle scFoundation de 100 millions de paramètres
Le modèle scFoundation développé par les chercheurs comporte environ 100 millions de paramètres, et son échelle de paramètres, sa couverture génétique et son échelle de données sont « parmi les meilleures » dans le domaine des cellules uniques.
Conception du modèle
Les chercheurs ont développé le modèle xTrimoGene comme modèle de base de scFoundation, qui est un modèle évolutif basé sur Transformer qui comprend un module d'intégration et une structure d'encodeur-décodeur asymétrique.
Parmi eux, le module vectoriel convertit les valeurs scalaires d'expression génétique continue en vecteurs de grande dimension pouvant être appris, garantissant que les valeurs d'expression d'origine sont entièrement préservées ; l'encodeur prend en entrée des gènes d'expression non nuls et non masqués, utilise un bloc de transformateur vanille et possède un grand nombre de paramètres ; le décodeur prend tous les gènes en entrée, utilise un bloc performer et possède un nombre relativement restreint de paramètres.
Cette conception asymétrique réduit les défis de calcul et de mémoire par rapport à d’autres architectures.Les données montrent que le module ne nécessite que 3,4% du modèle de langage traditionnel Transformer tout en conservant la même échelle de paramètres.
Tâches de pré-formation
Les chercheurs ont conçu une tâche de pré-formation appelée modélisation RDA (read-depth-aware).Il s’agit d’une extension du modèle de langage masqué qui prend en compte la grande variance de la profondeur de séquençage dans les données à grande échelle.
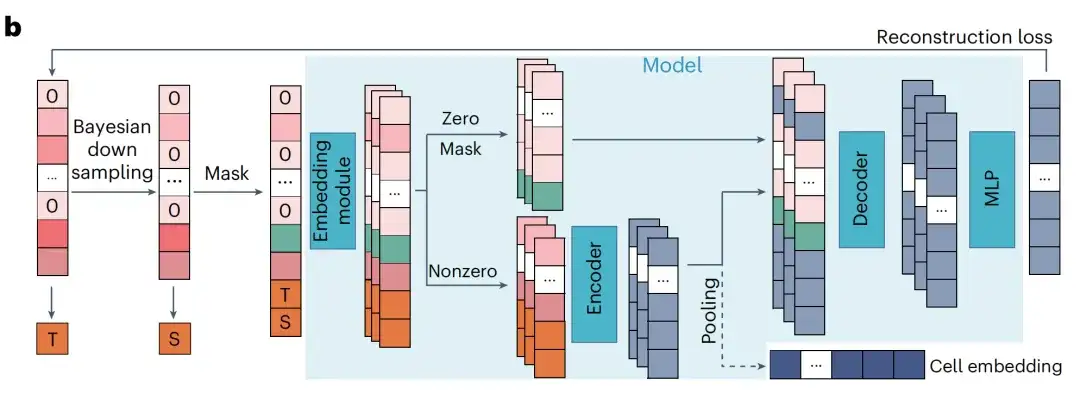
Dans la modélisation RDA, le modèle prédit l’expression des gènes masqués en fonction des gènes contextuels de la cellule. Les chercheurs ont considéré le nombre total comme la profondeur de séquençage d'une cellule et ont défini deux mesures de nombre total : T (cible) et S (source), qui représentent respectivement les nombres totaux de l'échantillon d'origine et de l'échantillon d'entrée. Les chercheurs ont masqué de manière aléatoire les gènes exprimés à zéro et non à zéro dans les échantillons d’entrée et ont enregistré leurs indices.
Le modèle utilise ensuite l’échantillon d’entrée masqué et les deux métriques pour prédire la valeur d’expression de l’échantillon d’origine à l’index masqué. Cela permet au modèle pré-entraîné non seulement de capturer les relations génétiques au sein des cellules, mais également de coordonner les cellules de différentes profondeurs de séquençage. Lors de l'inférence, les chercheurs saisissent l'expression génétique brute des cellules dans le modèle pré-entraîné et définissent T plus haut que leur nombre total S pour générer des valeurs d'expression génétique avec une profondeur de séquençage améliorée.
En termes simples, RDA peut sous-échantillonner la profondeur de séquençage, de sorte qu'en plus de terminer la tâche traditionnelle de récupération de masque, le modèle peut également récupérer les informations d'expression génétique des cellules de haute qualité à partir de cellules de faible qualité pendant la phase de pré-formation.
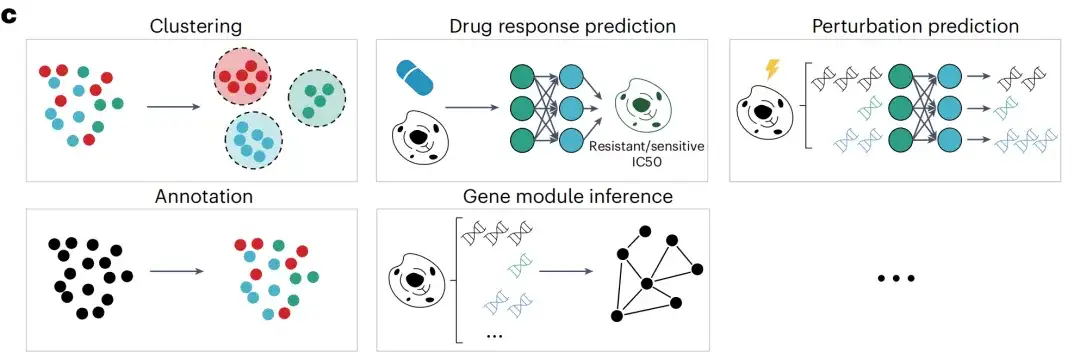
Après la pré-formation, les chercheurs ont ensuite appliqué le modèle scFoundation à plusieurs tâches en aval. Les sorties des encodeurs scFoundation sont résumées dans des vecteurs au niveau cellulaire pour être utilisées dans des tâches au niveau cellulaire, notamment le clustering (au sein et entre les ensembles de données), la prédiction de la réponse aux médicaments aux niveaux des lots et des cellules individuelles et l'annotation du type de cellule. La sortie du décodeur scFoundation est un vecteur de contexte au niveau des gènes, qui est utilisé pour les tâches au niveau des gènes telles que la prédiction des perturbations et l'inférence des modules génétiques.
Résultats de recherche : les modèles scFoundation ont d'excellentes performances
Dans les applications réelles, le modèle scFoundation prend en charge deux modes : « prêt à l'emploi » et « réglage fin ».En mode « prêt à l'emploi », grâce à ses tâches de pré-formation uniques, le modèle peut être directement utilisé pour améliorer la qualité des données cellulaires et peut obtenir des résultats identiques ou supérieurs aux méthodes existantes sans autre ajustement. De plus, les utilisateurs peuvent utiliser scFoundation pour extraire des représentations pré-entraînées de cellules, qui peuvent être utilisées pour identifier des modules génétiques et des facteurs de transcription spécifiques au type de cellule et peuvent être largement utilisés dans les tâches en aval.
Modèle d'amélioration profonde du séquençage évolutif et sans réglage fin
Les chercheurs ont formé trois modèles avec des paramètres de 3M, 10M et 100M respectivement et ont enregistré leurs pertes sur l'ensemble de données de validation.
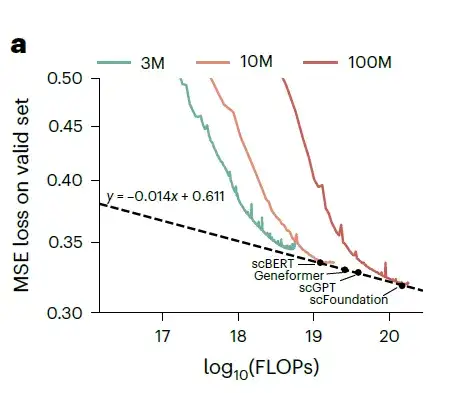
Perte d'entraînement sous différentes tailles de paramètres et FLOP. La courbe verte représente le modèle 3M, la courbe orange représente le modèle 10M et la courbe rouge représente le modèle 100M.
À mesure que les paramètres du modèle et les opérations en virgule flottante (FLOP) augmentent, la perte sur l'ensemble de données de validation montre une diminution de la loi de puissance. Les chercheurs ont ensuite estimé les performances des modèles d’architecture xTrimoGene de différentes tailles et les ont comparés à scVI. Comme le montre la figure ci-dessus,Le modèle scFoundation avec 100 millions de paramètres a obtenu les meilleurs résultats parmi tous les modèles.Les chercheurs ont ensuite évalué les trois modèles sur la tâche d’annotation du type de cellule et ont observé que les performances s’amélioraient à mesure que la taille du modèle augmentait.
Les chercheurs ont évalué cette capacité sur un ensemble de données de test indépendant de 10 000 cellules échantillonnées de manière aléatoire à partir de l'ensemble de données de validation, en sous-échantillonnant les nombres totaux à 1%, 5%, 10% et 20% des données d'origine, générant quatre ensembles de données avec différentes variations de nombre total. Pour chaque ensemble de données, l'erreur absolue moyenne (MAE), l'erreur relative moyenne (MRE) et le coefficient de corrélation de Pearson (PCC) entre les valeurs prédites et l'expression génétique réelle non nulle ont été mesurés à l'aide de scFoundation non réglé.
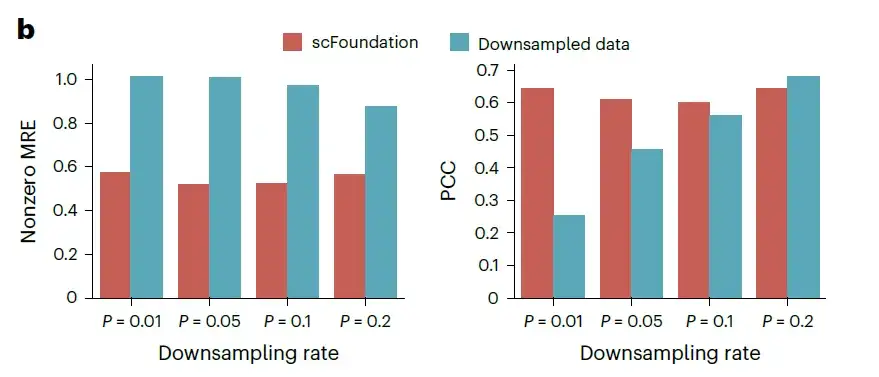
Évaluation des performances d'amélioration de la profondeur de lecture sur des ensembles de données inconnus MRE et PCC ont été utilisés pour évaluer les performances de l'expression génique récupérée, avec un MRE inférieur et un PCC supérieur indiquant de meilleures performances.
Comme le montre la figure ci-dessus, même lorsque le taux de sous-échantillonnage est inférieur à 10%, le MAE et le MRE de scFoundation sont considérablement réduits de moitié.Ces résultats démontrent la capacité de scFoundation à améliorer l’expression génétique à des nombres totaux extrêmement faibles.
Tâches en aval - tâches de prédiction de la réponse aux médicaments contre le cancer
Les réponses aux médicaments anticancéreux (CDR) visent à étudier les réponses des cellules tumorales à l'intervention médicamenteuse, et la prédiction informatique des CDR est cruciale pour guider la conception de médicaments anticancéreux et comprendre la biologie du cancer. Dans cette étude, les chercheurs ont combiné scFoundation avec la méthode de prédiction CDR DeepCDR pour prédire la valeur de la concentration inhibitrice demi-maximale IC50 des médicaments dans plusieurs données de lignées cellulaires afin de vérifier si scFoundation peut fournir des informations d'intégration utiles pour les données globales d'expression génétique basées sur une formation à cellule unique.
Les chercheurs ont évalué les performances des résultats basés sur scFoundation par rapport aux résultats basés sur l’expression génétique sur plusieurs médicaments et lignées cellulaires cancéreuses.Les résultats ont montré que la plupart des médicaments et tous les types de cancer obtenaient des coefficients de corrélation de Pearson (PCC) plus élevés en utilisant l'intégration scFoundation.Comme le montre la figure suivante :
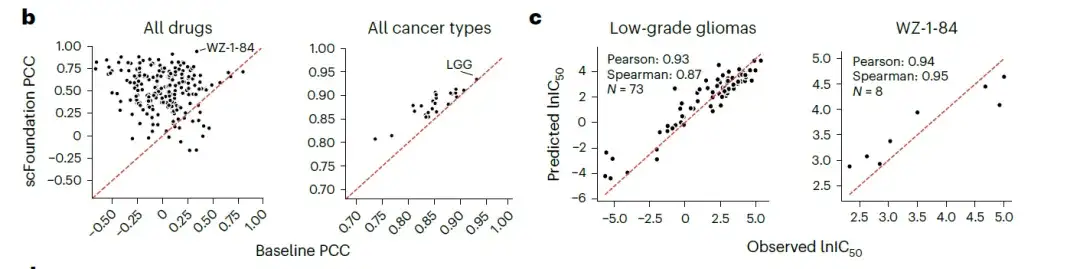
- Remarque : Le coefficient de corrélation de Pearson est une statistique qui mesure la force de la relation linéaire entre les variables, et sa plage de valeurs est comprise entre -1 et 1. Si le coefficient de corrélation est proche de 1, cela indique qu'il existe une relation linéaire complètement positive entre les deux variables ; s'il est proche de -1, cela indique qu'il existe une relation linéaire complètement négative ; s'il est proche de 0, cela indique qu'il n'y a pas de relation linéaire entre les deux variables.
Cela montre queBien que scFoundation soit pré-entraîné sur des données de transcriptome unicellulaire, les relations génétiques apprises peuvent être transférées aux données d'expression globales.Génère un vecteur compressé, facilitant des prédictions IC50 plus précises. Par conséquent, la scFoundation a le potentiel d’élargir notre compréhension des réponses aux médicaments dans la biologie du cancer et de guider la conception de traitements anticancéreux plus efficaces.
Tâche en aval - tâche de classification de la réponse aux médicaments d'une cellule unique
La déduction de la sensibilité aux médicaments au niveau d’une cellule unique peut aider à identifier des sous-types de cellules spécifiques qui présentent des profils de résistance aux médicaments distincts, fournissant des informations précieuses sur les mécanismes sous-jacents et les nouvelles approches thérapeutiques. Les chercheurs ont donc appliqué scFoundation à la tâche critique de classification de la réponse aux médicaments à cellule unique, sur la base d'un modèle en aval appelé SCAD.
Les chercheurs se sont concentrés sur quatre médicaments (sorafenib, NVP-TAE684, PLX4720 et étoposide) qui ont montré des valeurs d'ASC (aire sous la courbe) inférieures dans les études originales. Le modèle basé sur scFoundation a été comparé au modèle SCAD de base qui utilisait toutes les valeurs d'expression génétique comme entrée. Les résultats ont montré que le modèle basé sur scFoundation a obtenu des scores plus élevés dans les valeurs AUC de tous les médicaments, en particulier pour NVP-TAE684 et sorafenib, avec des valeurs AUC augmentées de plus de 0,2, comme le montre la figure ci-dessous.
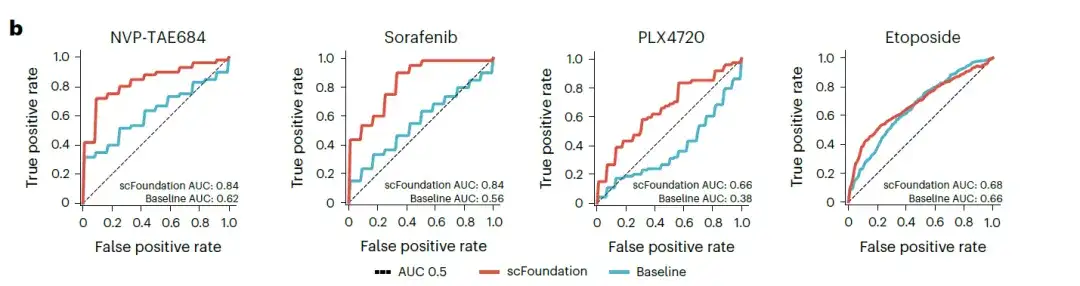
Remarque : l’AUC peut être utilisée pour mesurer les performances du modèle. La plage de valeurs de l'AUC est comprise entre 0 et 1. Plus la valeur est élevée, meilleures sont les performances de classification du modèle.
Ces résultats valident le potentiel de l’utilisation de l’intégration de scFoundation pour capturer les signaux des biomarqueurs de sensibilité aux médicaments.
Tâche en aval - tâche de prédiction de perturbation cellulaire
La compréhension des réponses cellulaires aux perturbations est essentielle pour les applications biomédicales et la conception de médicaments, car elle permet d’identifier les interactions gène-gène et les cibles médicamenteuses potentielles dans différents types de cellules. Les chercheurs ont combiné scFoundation avec un modèle avancé, GEARS, pour prédire les réponses aux perturbations à une résolution unicellulaire et ont calculé l'erreur quadratique moyenne (MSE) des 20 principaux gènes différentiellement exprimés (DE) avec des profils d'expression génétique significativement différents avant et après comme critère d'évaluation.
Les résultats montrent queComparé au modèle de base GEARS original, le modèle basé sur scFoundation a obtenu des valeurs MSE inférieures.La figure ci-dessous montre les changements d'expression des 20 principaux gènes dans la double perturbation génique ETS2 + CEBPE :
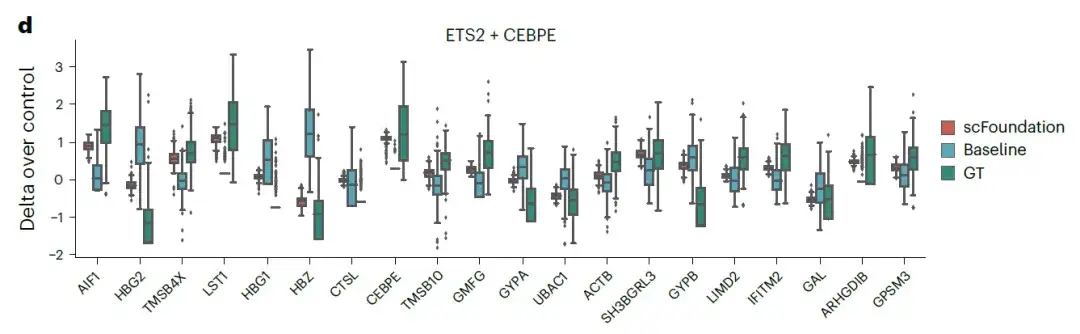
Ces résultats suggèrent qu’en extrayant des représentations génétiques à partir de cellules individuelles pour construire des réseaux spécifiques de co-expression génétique,scFoundation a réussi à capturer les caractéristiques cellulaires et génétiques dans différentes conditions et a considérablement amélioré la précision des prédictions de perturbations simples/doubles.
En résumé, le modèle scFoundation fournit de nouvelles idées et méthodes pour établir l'architecture du modèle, le cadre de formation et le système d'application de démonstration en aval des modèles de pré-formation à grandes cellules, fournit des fonctions de base pour l'apprentissage des tâches biomédicales et élargit les limites des modèles de base dans le domaine unicellulaire.
Explorer les grands modèles des sciences de la vie avec de meilleures performances
Demis Hassabis, PDG et fondateur de DeepMind, une société d'intelligence artificielle de premier plan au monde, a déclaré un jour :« À la base, la biologie peut être considérée comme un système de traitement de l'information très complexe et dynamique. Tout comme les mathématiques se sont révélées être le langage descriptif idéal pour la physique, la biologie pourrait être un domaine idéal pour les applications de l'intelligence artificielle. »
Cependant, les méthodes d’IA traditionnelles nécessitent de grandes quantités de données étiquetées pour faire des prédictions précises. Mais dans les sciences de la vie, les données étiquetées de haute qualité sont souvent rares. Vouloir créer des modèles de tâches en aval plus précis basés sur moins de données signifie que le modèle de base sous-jacent doit avoir une meilleure représentation ou des capacités générales. C’est pourquoi de plus en plus de chercheurs ont commencé à travailler à la conception de meilleurs modèles macro verticaux dans le domaine biologique.
Mai 2023Une équipe de recherche de l’Université de Toronto a publié le premier modèle linguistique à grande échelle basé sur la biologie unicellulaire, scGPT.Pré-entraîné sur plus de 10 millions de cellules, le modèle permet un apprentissage par transfert sur diverses tâches en aval. En juillet de la même année, l’équipe a tenté de mettre à jour le scGPT en générant une pré-formation sur plus de 33 millions de cellules. Les résultats ont montré que le scGPT peut extraire efficacement des informations biologiques clés sur les gènes et les cellules et atteindre des performances avancées dans diverses tâches en aval, notamment l'intégration multi-lots, l'intégration multi-omique, l'annotation du type de cellule, la prédiction des perturbations génétiques et l'inférence du réseau génétique.
L'étude, intitulée « scGPT : vers la construction d'un modèle de base pour la multi-omique unicellulaire à l'aide de l'IA générative », a été publiée dans Nature Methods.
* Lien vers l'article :https://www.nature.com/articles/s41592-024-02201-0
Septembre 2023Le consortium Xcompass, une équipe de recherche multidisciplinaire de l'Académie chinoise des sciences, a construit avec succès le premier modèle de fondation de vie inter-espèces au monde : GeneCompass.Le modèle intègre les données du transcriptome de plus de 126 millions de cellules individuelles provenant d'humains et de souris, fusionne quatre types de connaissances antérieures, notamment les séquences promotrices et les relations de co-expression des gènes, et dispose de 130 millions de paramètres de base du modèle, permettant un apprentissage et une compréhension panoramiques des lois de la régulation de l'expression des gènes, tout en soutenant la prédiction des changements d'état cellulaire et l'analyse précise de divers processus vitaux.

L'étude a été publiée sur bioRxiv sous le titre « GeneCompass : déchiffrer les mécanismes universels de régulation des gènes avec un modèle de fondation inter-espèces basé sur les connaissances ».
- Lien vers l'article :https://www.biorxiv.org/content/10.1101/2023.09.26.559542v1
En octobre 2023, le géant pharmaceutique mondial Sanofi a annoncé un partenariat stratégique à grande échelle avec BioMap BioScience. Les deux parties développeront conjointement des modèles de pointe pour la découverte de médicaments biothérapeutiques basés sur le modèle Life Science AI Foundation de BioMap.
En regardant vers l’avenir, l’application de la compréhension complexe et des capacités de génération innovantes de grands modèles de langage qui dépassent de loin l’imagination humaine au « langage naturel » plus complexe de la vie changera, espérons-le, véritablement le paradigme de la recherche en sciences de la vie.
Références :
1.https://www.jiqizhixin.com/articles/2023-9-29
2.https://www.tsinghua.edu.cn/info/1175/112118.htm
3.https://hope.huanqiu.com/article/4FYZxnpu88J
4.https://www.jiqizhixin.com/articles/2023-7-5-26'








