Command Palette
Search for a command to run...
Nouvelle Réalisation De l'Institut Fudan Des Sciences Du Cerveau : Utiliser La Segmentation Sémantique Comme Référence Pour Développer Un Outil d'annotation Sémantique Du Transcriptome Spatial Pianno

Depuis qu'elle a été sélectionnée comme technologie de l'année par Nature Methods en 2020,La « transcriptomique spatiale » est devenue l’une des technologies révolutionnaires les plus en vogue dans le domaine des sciences de la vie aujourd’hui.En termes simples, cette technologie peut obtenir des informations spatiales sur les tissus et des données de transcriptome, analyser avec précision les modèles d’expression génétique dans les tissus, ainsi que les caractéristiques biologiques telles que la relation de position spatiale des populations cellulaires à partir des dimensions temporelles et spatiales. Il est d’une grande valeur dans la recherche dans les domaines de la recherche sur les maladies, de la croissance et du développement, de la structure des organes et de l’évolution des espèces.
Alors que la transcriptomique spatiale continue d'être populaire dans la recherche universitaire, des technologies de transcriptomique spatiale telles que 10x Visium, Slide-seq et Stereo-seq ont également émergé. Ces dernières réalisations et avancées bouleversent complètement la recherche humaine sur les modèles d’expression génétique dans les tissus. Cependant, la simple obtention du profil d’expression génétique à des coordonnées physiques spécifiques au sein d’un tissu ne suffit pas à comprendre pleinement la complexité des systèmes biologiques. Pour comprendre pleinement la complexité, il est nécessaire d’identifier l’identité biologique de chaque point spatial au sein du tissu.
Actuellement, les méthodes basées sur l’apprentissage automatique sont largement utilisées pour identifier des groupes de points spatiaux et interpréter leurs identités biologiques à l’aide de gènes marqueurs.Cependant, ces méthodes sont souvent limitées par leur incapacité à établir des connexions explicites avec des structures connues au sein du cluster.De plus, l’annotation manuelle est souvent utilisée pour aider à identifier les structures connues, mais cette méthode est souvent limitée par l’expertise et le jugement subjectif des chercheurs et ne peut pas être appliquée à une analyse à grande échelle.
En réponse aux défis ci-dessus, l'équipe de Zhu Ying de l'Institut des sciences du cerveau de l'Université Fudan a récemment publié un résultat de recherche intitulé « Pianno : un cadre probabiliste automatisant l'annotation sémantique pour la transcriptomique spatiale » dans « Nature Communications ».L'équipe de recherche a emprunté l'idée de « segmentation sémantique » à la vision par ordinateur, a proposé le concept d'« annotation sémantique du transcriptome spatial » et a développé l'outil d'annotation sémantique du transcriptome spatial Pianno.La capacité de définir automatiquement des structures ou des types de cellules pour des points spatiaux dans les tissus peut combiner des informations provenant de plusieurs dimensions pour améliorer l'interprétation de systèmes biologiques complexes.
Points saillants de la recherche :
* Pianno dispose d'un mode d'étiquetage automatique unique qui s'applique aux données générées par diverses technologies de transcriptomique spatiale
* Pianno démontre des performances supérieures par rapport aux méthodes de clustering spatial de pointe, offrant de nouvelles perspectives sur les données transcriptomiques spatiales

Adresse du document :
https://doi.org/10.1038/s41467-024-47152-4
Ensemble de données : données publiques, calculs rigoureux
Les ensembles de données utilisés dans cette étude sont principalement des ensembles de données publics provenant de différentes plateformes de technologie spatiale, notamment l'ensemble de données du cortex préfrontal dorsolatéral humain dlPFC, l'ensemble de données de section coronale de l'hémisphère cérébral de souris adulte Stereo-seq, l'ensemble de données de prétraitement de l'hippocampe de souris Slide-seqV2, l'ensemble de données d'adénocarcinome canalaire pancréatique humain ST, l'ensemble de données de cancer du sein humain Visium, l'ensemble de données du cortex visuel primaire de souris scRNA-seq, les ensembles de données snRNA-seq de plusieurs régions corticales humaines et les images de coloration DAPI du bulbe olfactif de la souris.
Dans l'étude, afin d'éviter la destruction des caractéristiques biologiques originales par des technologies de traitement d'image telles que la réduction du bruit, le lissage et la netteté,L’équipe de recherche a construit un classificateur bayésien basé sur les décomptes bruts pour affiner les annotations initiales.Dans le même temps, l’équipe de recherche a appliqué un modèle a priori de champ aléatoire de Markov (MRF) d’ordre élevé. Dans le contexte de la transcriptomique spatiale, étant donné que l’expression génétique et la position spatiale de chaque site doivent être considérées ensemble, l’équipe de recherche a également adopté le modèle de processus ponctuel spatial de Poisson (sPPP).
Pianno : Nouvel outil innovant pour l'annotation sémantique automatisée du transcriptome spatial
L’équipe de recherche a proposé un nouvel outil appelé Pianno basé sur le cadre bayésien.L'outil combine les champs aléatoires de Markov (MRF) avec les processus ponctuels spatiaux de Poisson (sPPP), exploitant pleinement la capacité des sPPP à modéliser la distribution des données de comptage d'ARN-seq tout en prenant en compte les informations de localisation des points spatiaux. Il peut annoter automatiquement l’identité biologique de chaque point dans les données du transcriptome spatial à l’aide d’une liste prédéfinie de gènes marqueurs.
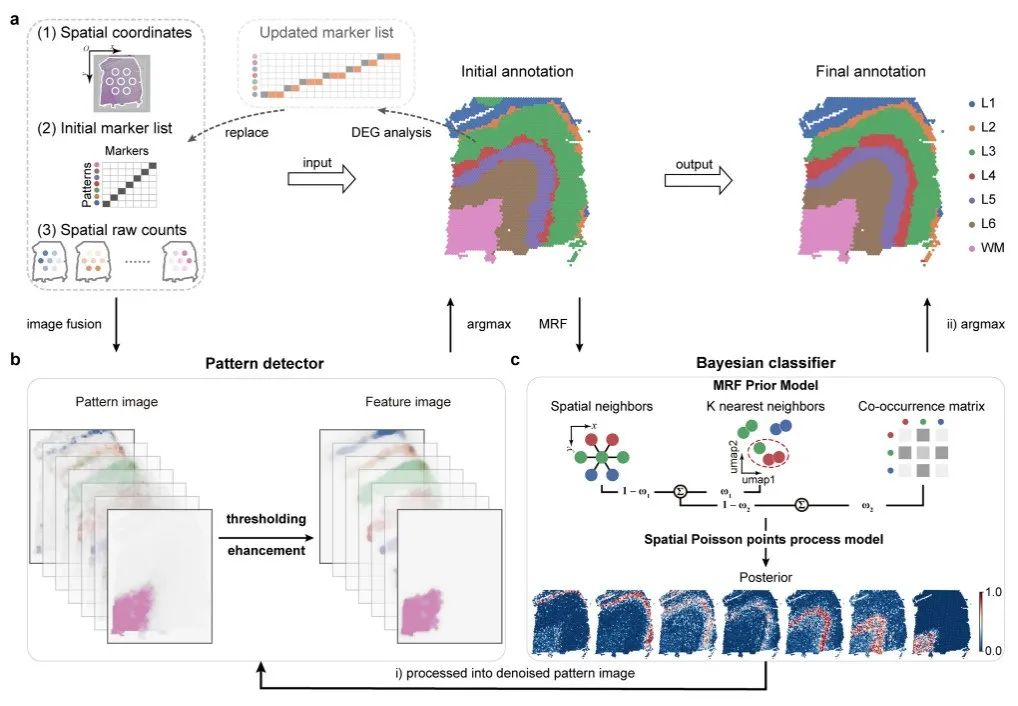
Les données de transcriptome spatial saisies par Pianno comprennent les coordonnées spatiales, la liste initiale des gènes marqueurs et le nombre brut de gènes.Chaque modèle fournit au moins un jeton connu.
Le processus d'annotation comprend une étape de segmentation initiale et une étape de raffinement :
Dans l’étape de segmentation initiale,L’expression spatiale de chaque gène a été convertie en une image en niveaux de gris. Pour chaque modèle cible, une image de modèle est créée en agrégeant les images en niveaux de gris des gènes marqueurs associés à ce modèle, puis des gènes marqueurs candidats supplémentaires pour chaque modèle sont déterminés pour mettre à jour la liste de marqueurs initiale. Les listes de marqueurs mises à jour seront intégrées dans les étapes de raffinement ultérieures, en tenant compte de leurs modèles d'expression uniques dans les structures initialement annotées.
Dans l'étape de raffinement,Un classificateur bayésien est construit pour évaluer la probabilité postérieure que chaque point spatial appartienne à différents modes, puis l'annotation est mise à jour en fonction de la probabilité postérieure.
Pianno propose deux méthodes pour mettre à jour les annotations :
* Pour les modèles continus dans l'annotation sémantique, il est recommandé d'utiliser la distribution de probabilité comme image de modèle et de la renvoyer au détecteur de modèle pour mettre à jour l'annotation ; * Pour les motifs d'image dispersés ou nets, il est recommandé de mettre à jour l'étiquette directement en fonction de la valeur de probabilité car elle peut conserver des informations détaillées.
En général,Pianno a simplifié le processus d'annotation et adopté une approche heuristique pour utiliser un seul gène marqueur initial pour identifier des gènes marqueurs supplémentaires, ce qui peut minimiser l'entrée du nombre de marqueurs connus.
Résultats de la recherche : excellentes performances et forte applicabilité
Dans cette étude, l’équipe de recherche a vérifié les performances, la précision et l’adaptabilité de Pianno, et a démontré davantage les capacités de Pianno en le comparant aux méthodes existantes.
Dans une comparaison avec des outils basés sur le clustering pour l'annotation de structures anatomiques, l'équipe de recherche a évalué les performances de Pianno à l'aide de 12 échantillons de l'ensemble de données dlPFC et les a comparées à CellAssign, une autre méthode d'annotation basée sur des marqueurs mais sans informations spatiales. De plus, l'algorithme de clustering non supervisé de Leiden et cinq méthodes de clustering spatial (SpaGCN, SEDR, BayesSpace, DeepST et STAGATE) ont également été pris en compte dans le processus d'évaluation.
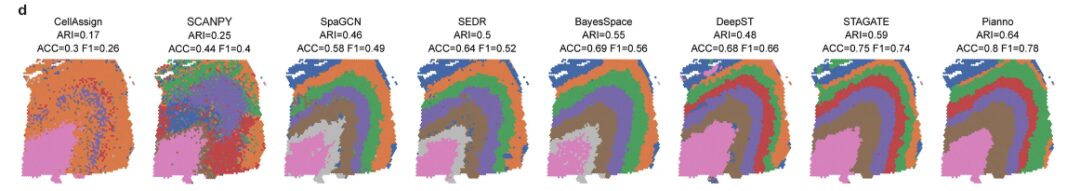
L’évaluation a révélé queLes performances de Pianno ont obtenu la plus grande concordance avec les annotations manuelles réalisées par des chercheurs expérimentés sur la base de caractéristiques morphologiques et de marqueurs.Onze des douze échantillons ont surpassé les autres méthodes de test.
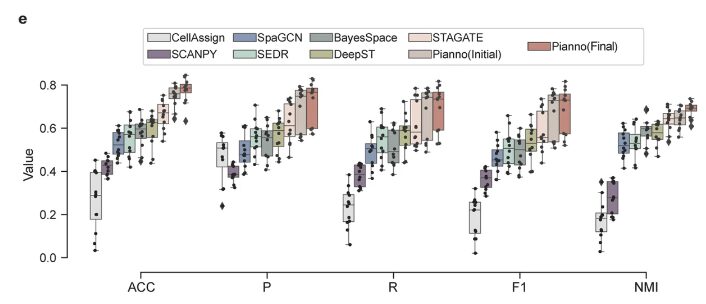
En outre, l'équipe de recherche a évalué de manière plus approfondie les performances supérieures de Pianno à l'aide d'autres indicateurs de classification, tels que la précision (ACC), la précision de la macro-moyenne (P), le rappel de la macro-moyenne (R), le score F1 de la macro-moyenne (F1) et l'information mutuelle normalisée (NMI), comme le montre la figure e ci-dessus.Les indicateurs liés au piano sont tous à un niveau élevé.
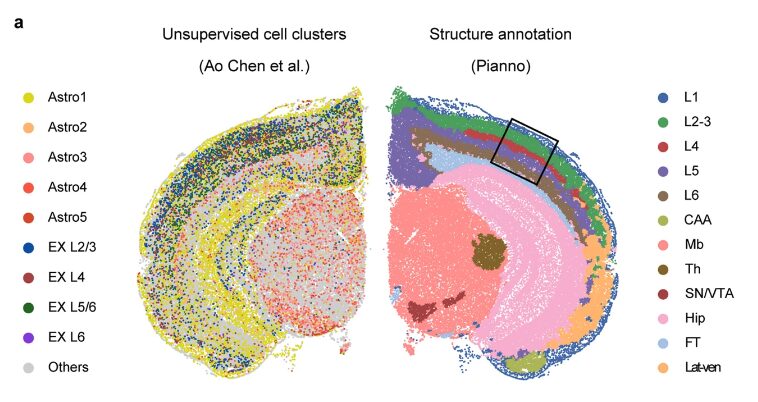
L’équipe de recherche a ensuite évalué la capacité de Pianno à prédire la distribution spatiale des types de cellules. Dans cette série de validation, l'équipe de recherche a utilisé un ensemble de données Stereo-seq de sections coronales d'hémicerveau de souris adulte et a comparé les résultats avec la distribution des types de cellules déduite par différentes stratégies, notamment le clustering non supervisé après segmentation cellulaire et trois outils de déconvolution spatiale basés sur l'intégration de la transcriptomique spatiale et unicellulaire.
L'étude a révélé que les prédictions de Pianno concernant la distribution des sous-types de neurones excitateurs présentaient des modèles comparables à ceux de Tangram et de RCTD, et étaient très cohérentes avec leurs emplacements connus dans chaque couche. En général,Ces résultats démontrent la robustesse et la précision de Pianno dans la prédiction de distributions complexes de types de cellules dans des ensembles de données spatiales, en particulier dans les situations où les méthodes non supervisées sont confrontées à des défis.
L'équipe de recherche a ensuite évalué plus en détail les performances de Pianno dans l'annotation de diverses structures de forme dans les données de transcriptome spatial sur différentes plates-formes et les a comparées à STAGATE.
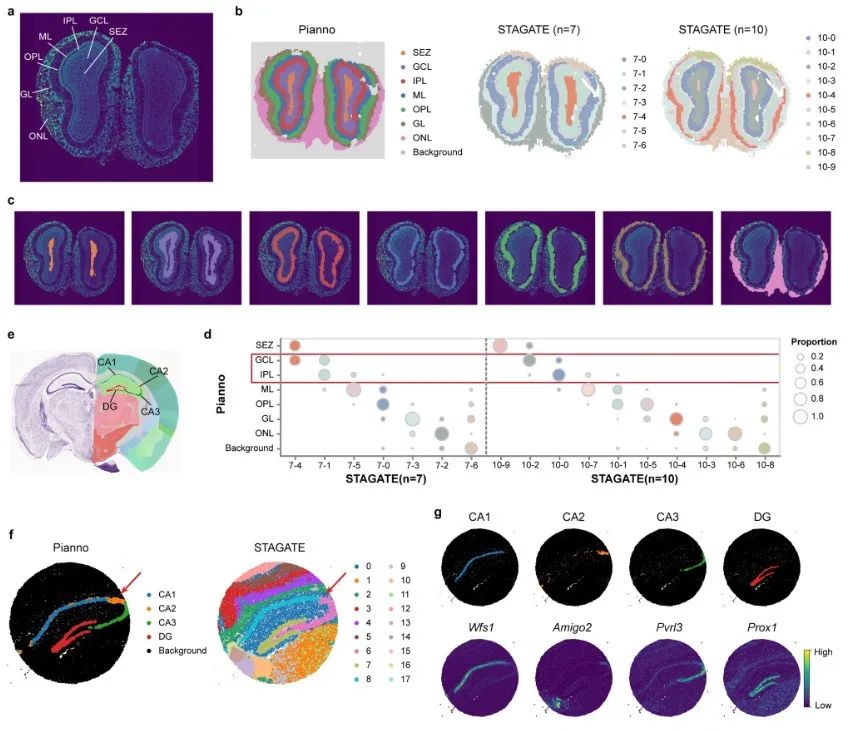
L'équipe de recherche a utilisé Pianno pour annoter les structures anatomiques dans l'ensemble de données Stereo-seq du bulbe olfactif de la souris, qui contient 10 747 points spatiaux couvrant à la fois les zones recouvertes de tissu et les zones d'arrière-plan.
Pianno a pu effectuer simultanément une soustraction d'arrière-plan et une annotation de structure en quelques minutes. En revanche, lorsque le nombre de clusters est défini sur le nombre de structures, STAGATE ne parvient pas à identifier les clusters correspondant à toutes les structures anatomiques.
L'équipe de recherche a également évalué les performances de Pianno dans l'annotation d'organisations structurelles complexes et dispersées à la lumière de la forte hétérogénéité du microenvironnement tumoral. Cette série de tests a analysé le microenvironnement de deux échantillons d’adénocarcinome canalaire pancréatique humain et de deux échantillons de cancer du sein.
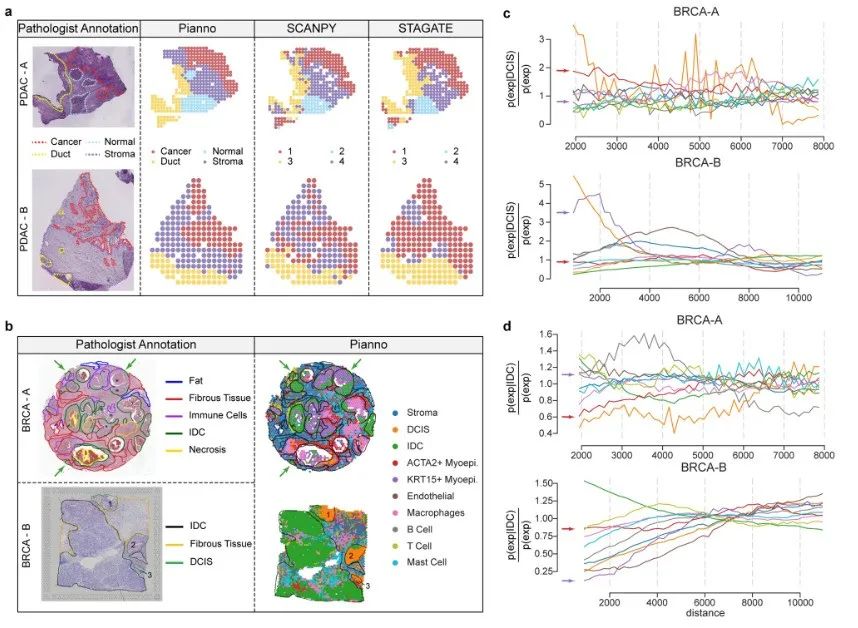
Dans l'ensemble,Pianno a montré un niveau de cohérence avec l'annotation manuelle par des pathologistes professionnels, démontrant son grand potentiel dans l'annotation de structures irrégulières et complexes, en particulier dans des microenvironnements tumoraux hétérogènes.Cela fournit une aide précieuse aux pathologistes pour comprendre la complexité de la biologie tumorale et devrait fournir de nouvelles idées pour fournir des stratégies de traitement personnalisées.
Combiner l’intelligence artificielle avec la biologie complexe a un grand potentiel
Selon l'Institut des sciences du cerveau de l'Université Fudan, le projet de recherche a été financé par le projet clé du programme national de R&D clé « Intégration biologique et informationnelle (intégration BT et IT) », le projet majeur d'innovation scientifique et technologique 2030 - « Recherche sur les sciences du cerveau et les sciences du cerveau », la Fondation nationale des sciences naturelles, le projet majeur de science et technologie de Shanghai et le laboratoire de Zhangjiang.
Il est entendu que l'Institut des sciences du cerveau de l'Université Fudan a été créé en avril 2006. Il s'agit d'une entité de recherche en neurosciences à l'échelle de l'école de l'Université Fudan et l'une des principales plateformes d'innovation scientifique et technologique construites dans la deuxième phase du « Projet 985 » du ministère de l'Éducation. Il s'agit d'un projet de construction « deux en un » avec le Laboratoire national clé de neurobiologie médicale.
Depuis sa création, l'Institut des sciences du cerveau de l'Université Fudan a obtenu des résultats fructueux. L'institut a répondu à maintes reprises à des besoins internationaux et nationaux majeurs, entrepris des projets de recherche scientifique majeurs et produit des résultats de recherche importants. Selon son site officiel, les chercheurs de l'institut ont présidé et participé à une série de projets de recherche scientifique majeurs, notamment le « Programme 973 » du ministère des Sciences et de la Technologie, le « Programme 863 », l'Innovation scientifique et technologique 2030 « Recherche sur les sciences du cerveau et les sciences similaires au cerveau », le Programme national clé de R&D, le projet spécial national majeur de science et de technologie « Création majeure de nouveaux médicaments », etc.
En fait, outre l’Institut des sciences du cerveau de l’Université Fudan, de nombreux laboratoires et entreprises ont également commencé à s’intéresser à la technologie du transcriptome spatial.
Par exemple,L'équipe de Zhang Shihua de l'Institut de mathématiques et de sciences des systèmes de l'Académie chinoise des sciences a développé la série d'outils STA. En 2022, l'équipe a lancé STAGATE, un outil d'intelligence artificielle permettant d'identifier les sous-structures spatiales des tissus biologiques, adaptable à différentes technologies de transcriptome spatial et à différents tissus biologiques. Depuis son entrée en 2023, l'équipe a publié un certain nombre de résultats autour de la technologie du transcriptome spatial - * Mise en place d'un nouvel outil d'analyse intégré STAligner pour les données de transcriptome spatial multi-tranches de tissus biologiques provenant de différentes technologies, de différents moments de développement et de différentes pathologies. * STAMarker, une méthode d'identification de gènes variables spécifiques au domaine spatial basée sur des cartes de saillance d'apprentissage profond, réalise simultanément l'identification du domaine spatial et l'identification des gènes variables spatiaux correspondants, et devrait fournir une méthode efficace pour l'analyse fine des données du transcriptome spatial. * En collaboration avec l'équipe de Yang Yungui et Cai Jun de l'Institut de génomique de Pékin de l'Académie chinoise des sciences (Centre national de bioinformatique), nous avons cartographié la carte tridimensionnelle du transcriptome spatial STAPR du ver méditerranéen pendant la régénération et identifié systématiquement plusieurs facteurs régulateurs clés pour la régénération.

Le groupe de recherche du professeur Zhang Xiaofei à l'École de mathématiques et de statistiques de l'Université normale de Chine centrale a développé une méthode de calcul appelée ENGEP.En utilisant des stratégies de régression pondérée des k plus proches voisins et d’apprentissage d’ensemble, nous avons pu prédire avec précision l’expression de gènes non mesurés dans le transcriptome spatial. De plus, l’ENGEP peut prédire avec précision les modèles d’expression de gènes non mesurés spatialement, ce qui est d’une grande importance pour améliorer les données transcriptomiques spatiales.
Il ne fait aucun doute que l’autonomisation de l’IA dans la transcriptomique spatiale et même dans la biologie a non seulement amélioré l’efficacité de la recherche, mais a également fourni de nouvelles solutions aux difficultés de la recherche scientifique. Comme indiqué dans la section de discussion de l'article, la valeur apportée par Pianno est qu'il peut remplacer l'annotation manuelle à forte intensité de main-d'œuvre existante et fournir des formulaires efficaces, précis et peu coûteux de manière automatisée pour apporter des changements à la transcriptomique spatiale et promouvoir de nouveaux développements en biologie.
Références :
1. https://news.fudan.edu.cn/2024/0407/c2474a139894/page.htm
2. https://bfse.cas.cn/sxyqyjc/kyjz/202311/t20231110_4985132.html
3. https://kjc.ccnu.edu.cn/info/1009/3744.htm








