Command Palette
Search for a command to run...
Recherche Et Développement Indépendants ! L'équipe De l'Institut De Recherche Médicale Militaire a Proposé MIDAS, Qui Peut Être Utilisé Pour l'intégration En Mosaïque De Données multi-omiques Unicellulaires
Comme nous le savons tous, les cellules sont les plus petits éléments constitutifs de la vie. Le corps humain contient 40 à 60 billions de cellules, qui constituent la base de notre croissance et de notre développement. La réalisation de recherches au niveau de la cellule unique est essentielle pour comprendre avec précision la croissance et le développement cellulaires ainsi que le diagnostic et le traitement des maladies.
Ces dernières années, la technologie de séquençage de cellules uniques est devenue un sujet brûlant dans la recherche en biologie moléculaire. L’industrie a généré une grande quantité de données de séquençage de cellules uniques autour de questions de recherche clinique et fondamentale telles que la maladie et le développement. Cependant, les données massives provenant de différentes combinaisons omiques, de différentes technologies de séquençage et de différents échantillons de séquençage sont aussi dispersées et diverses que des carreaux de mosaïque sur le sol.Comment intégrer des données aussi volumineuses et désordonnées et mener des recherches biomédicales est un défi commun auquel sont confrontés les scientifiques du monde entier.
Afin de surmonter ce défi, l'équipe de Ying Xiaomin et l'équipe de Bo Xiaochen de l'Institut de recherche médicale militaire ont récemment mené Nature Biotechnologie La revue a publié un article de recherche intitulé « Intégration de mosaïque et transfert de connaissances de données multimodales unicellulaires avec MIDAS ».Cette étude a proposé un outil informatique MIDAS pour l'intégration en mosaïque de données omiques multimodales unicellulaires (scMulti-omics) (c'est-à-dire que différents ensembles de données ne partagent que certaines modalités de détection) et le transfert de connaissances.Sur la base d'approches d'apprentissage auto-supervisé et de théorie de l'information, nous avons réalisé pour la première fois les fonctions d'intégration universelles de données mosaïques multi-omiques unicellulaires, telles que l'alignement modal, la complétion des données et la correction par lots, fournissant des technologies originales importantes pour la construction de cartes de cellules multi-omiques à grande échelle et la réalisation d'analyses multi-omiques unicellulaires à grande échelle et de transfert de connaissances.
Points saillants de la recherche :
* Développé indépendamment un nouvel algorithme basé sur l'intelligence artificielle générative, MIDAS
* Pour la première fois, les fonctions d'intégration d'alignement de modalité, de complétion de données, de correction par lots, etc. de données mosaïques multi-omiques unicellulaires communes ont été réalisées.
* Le nouvel algorithme est d'une grande importance pour révéler les fonctions des cellules et les mécanismes de régulation moléculaire et pour étudier l'apparition et le développement des maladies

Adresse du document :
https://www.nature.com/articles/s41587-023-02040-y
Suivez le compte officiel et répondez « cellule unique » pour obtenir le PDF complet
Ensemble de données : Ensembles de données multiples, performances d'évaluation multidimensionnelles
Afin de comparer les avantages du modèle MIDAS à partir de différentes dimensions, cette étude a construit plusieurs ensembles de données.
Tout d’abord, pour comparer MIDAS avec les méthodes de pointe,Cette étude a évalué les performances de MIDAS dans l'intégration trimodale avec des modes complets (une forme simplifiée d'intégration mosaïque), une tâche que l'équipe de recherche a nommée « intégration rectangulaire ». L'équipe a utilisé deux souches humaines trimodales unicellulaires publiées PBMC ensembles de données (DOGMA-seq et TEA-seq), mesurant simultanément l'ARN, l'ADT et l'ATAC dans chaque cellule, construisant ainsi les ensembles de données dogma-full et teadog-full. Remarque : PBMC signifie cellule mononucléaire du sang périphérique, couramment utilisée dans les activités de recherche scientifique dans le domaine de l'immunologie.
Deuxièmement, pour évaluer les performances de MIDAS dans l’intégration de mosaïque,Sur la base de l'ensemble de données rectangulaire généré précédemment, l'équipe de recherche a construit 14 ensembles de données incomplets, chacun d'entre eux ayant été généré en supprimant plusieurs blocs de lots modaux de l'ensemble de données modal complet.
Troisièmement, afin d’étudier la capacité de transfert de connaissances de MIDAS,L’équipe de recherche a redivisé l’ensemble de données de l’atlas en un ensemble de données de référence utilisé pour la construction de l’atlas et un ensemble de données de requête. L'équipe de recherche a obtenu un ensemble de données de référence nommé atlas-no_dogma en supprimant DOGMA-seq de l'atlas.
Quatrièmement, étudier l’application de MIDAS dans des ensembles de données à cellule unique avec des changements continus d’état cellulaire,L'équipe de recherche a construit un ensemble de données mosaïques BMMC humaines en combinant trois échantillons différents (ICA, ASAP et CITE) obtenus à partir du scRNA-seq public (séquençage d'ARN à cellule unique).
Architecture du modèle : Modèle génératif profond MIDAS
MIDAS est un modèle génératif profond qui représente la distribution conjointe de données multimodales unicellulaires incomplètes, qui comprend des mesures de la chromatine accessible à la transposase (ATAC), de l'ARN et des étiquettes dérivées d'anticorps (ADT).
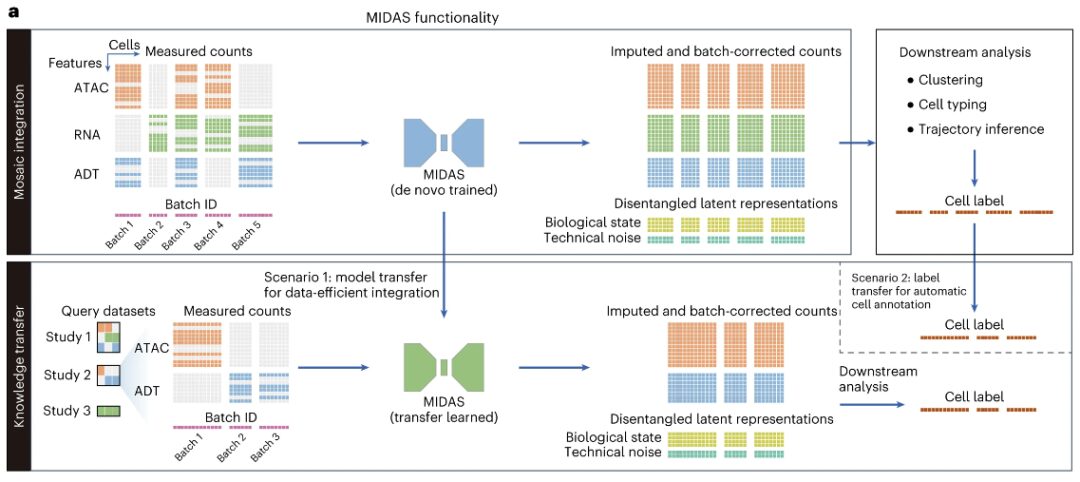
Plus précisément, MIDAS suppose que les mesures multimodales de chaque cellule sont générées via deux variables latentes indépendantes de la modalité et découplées (état biologique et bruit technique) basées sur un réseau neuronal profond.Son entrée comprend une matrice de comptage de cellules de caractéristiques en mosaïque composée de différents échantillons de cellules uniques (lots) et d'un vecteur représentant l'ID du lot de cellules.Ces échantillons unicellulaires peuvent provenir de différentes expériences ou être générés en appliquant différentes technologies de séquençage (telles que scRNA-seq, CITE-seq, ASAP-seq et TEA-seq), et peuvent donc avoir un bruit technique, des modalités et des caractéristiques différents.
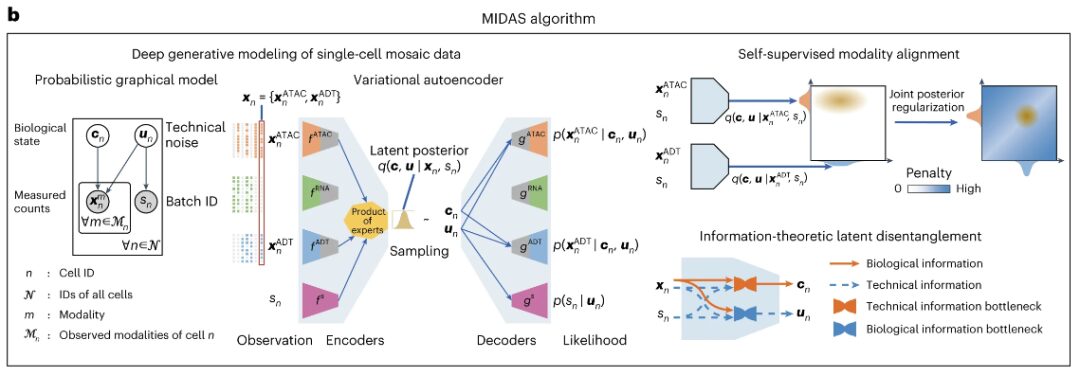
Les sorties de MIDAS comprennent des matrices d'état biologique et de bruit technique, ainsi que des matrices de comptage estimées et corrigées par lots, à partir desquelles les modalités et caractéristiques manquantes dans les données d'entrée sont interpolées et les effets de lots sont supprimés.Ces sorties peuvent être utilisées pour des analyses en aval telles que le clustering, la délimitation des types de cellules et l'inférence de trajectoire.
MIDAS est basé sur l'architecture d'autoencodeur variationnel (VAE) et dispose d'un réseau d'encodeurs modulaires et d'un réseau de décodeurs. Le premier peut traiter des données d'entrée en mosaïque et déduire des variables latentes, et le second peut utiliser des variables latentes pour démarrer le processus de génération de données observées. MIDAS utilise l'apprentissage auto-supervisé pour aligner différentes modalités dans l'espace latent, améliorant ainsi l'inférence intermodale dans les tâches en aval telles que l'interpolation et la traduction. Des méthodes théoriques de l’information sont également appliquées pour découpler l’état biologique et le bruit technique afin d’obtenir une correction par lots supplémentaire.
Les chercheurs ont combiné ces éléments dans les objectifs d'optimisation de cette étude et ont obtenu un apprentissage et une inférence évolutifs de MIDAS grâce à la méthode Bayes variationnelle à gradient stochastique (SGVB), ce qui a également rendu possible l'intégration de mosaïques à grande échelle et la construction de cartes de données multimodales à cellule unique. De plus, afin de transférer les connaissances de l'atlas construit pour interroger des ensembles de données avec différentes combinaisons de modalités, les chercheurs ont développé des schémas d'apprentissage par transfert et de cartographie de références croisées pour le transfert des paramètres du modèle et des étiquettes de cellules, respectivement.
Résultats de recherche : MIDAS est polyvalent et efficace
Les résultats de cette étude indiquent que MIDAS est un outil d’intégration multimodale monocellulaire puissant, polyvalent et efficace.
L’équipe de recherche a comparé les performances de MIDAS avec neuf méthodes récemment publiées en termes d’élimination des effets de lot et de préservation des signaux biologiques.
Les résultats montrent queMIDAS élimine idéalement les effets de lot et préserve les informations de type de cellule sur les ensembles de données dogma-full et teadog-full, tandis que les performances des autres méthodes sont légèrement inférieures.Par exemple, BBKNN+average, MOFA+, PCA+WNN, Scanorama-embed+WNN et Scanorama-feat+WNN n'ont pas bien mélangé les différents lots, et les groupes de cellules générés par PCA+WNN et Scanorama-feat+WNN étaient largement incompatibles avec les types de cellules.
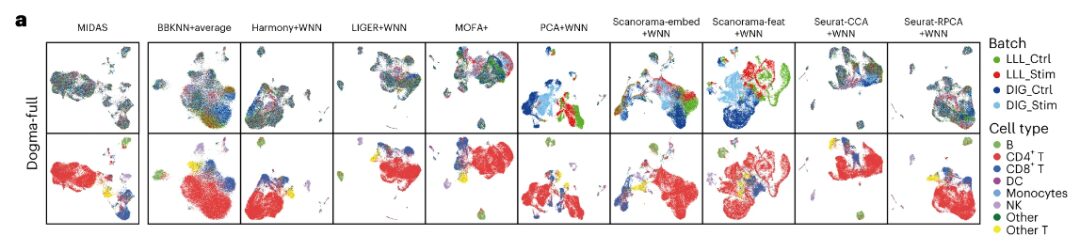
Résultats obtenus à partir de l'évaluation et de l'analyse en aval
En termes d'alignement des lots, MIDAS est capable d'aligner très bien les cellules de différents lots et de les regrouper de manière cohérente avec les étiquettes de type de cellule.D’autres méthodes ne mélangent pas bien les différents lots de cellules et produisent des groupes qui sont largement incompatibles avec le type de cellule. Le benchmark scIB montre que MIDAS a des performances stables sur différentes tâches de mosaïque et son score global est bien supérieur à celui des autres méthodes.
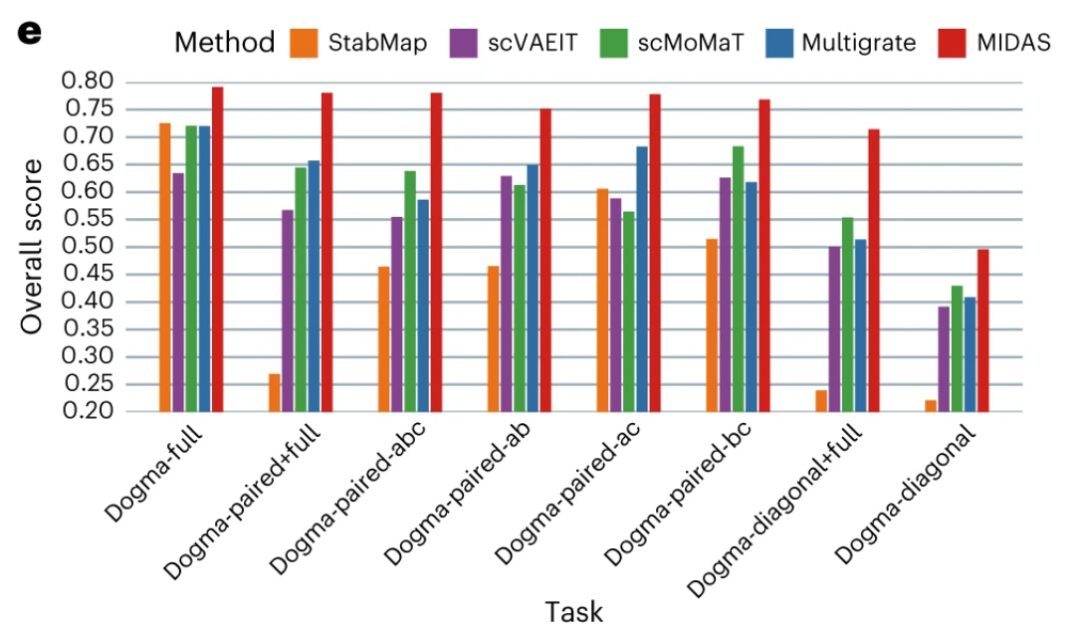
Scores d'évaluation de performance qualitatifs et quantitatifs
En termes de capacité de transfert de connaissances, les chercheurs ont aligné chaque ensemble de données de requête avec l'ensemble de données de référence et ont utilisé les k plus proches voisins (kNN) algorithme pour transférer les étiquettes de type de cellule. Après avoir cartographié et visualisé les états biologiques, on peut constater que les résultats de cartographie de référence croisée de différents ensembles de données de requête sont cohérents et hautement cohérents avec les résultats d'intégration de carte obtenus via l'ensemble de données dogmatique. MIDAS permet un transfert d'étiquettes robuste et précis, évitant ainsi le besoin d'une intégration de novo et d'une analyse en aval.Par conséquent, MIDAS peut être utilisé pour transférer des connaissances au niveau de l'atlas vers diverses formes d'ensembles de données utilisateur sans coûts de formation de novo coûteux ni analyse en aval complexe.
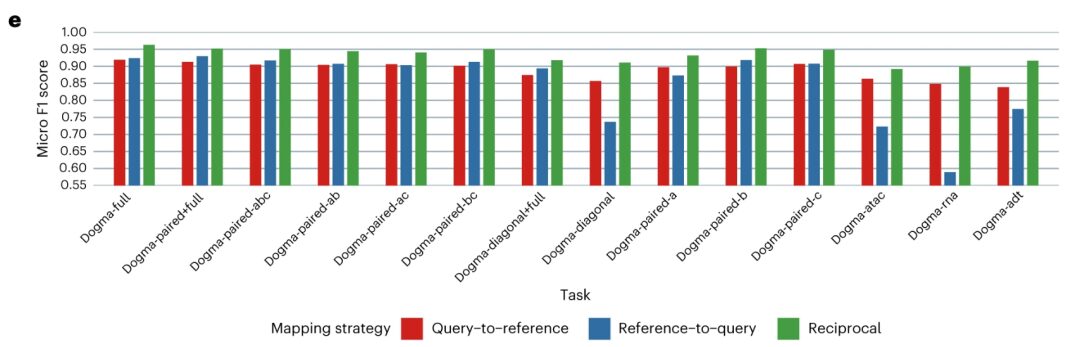
En résumé, en modélisant le processus de génération de données de mosaïque unicellulaire, MIDAS peut séparer avec précision les états biologiques et le bruit technique des entrées et ajuster de manière robuste les modalités pour prendre en charge les analyses intégrées multi-sources et hétérogènes. MIDAS fournit des résultats précis et robustes et surpasse les autres méthodes lors de l'exécution de diverses tâches d'intégration de mosaïque.
De plus, MIDAS transfère de manière efficace et flexible les connaissances des ensembles de données de référence aux ensembles de données de requête, facilitant ainsi le traitement de nouvelles données multi-omiques. Avec d'excellentes performances de réduction de dimensionnalité et de correction par lots, MIDAS prend en charge une bioanalyse précise en aval. En plus de permettre le regroupement et l'identification du type de cellule des données mosaïques, MIDAS peut également aider à l'analyse pseudo-temporelle des cellules avec des états séquentiels, ce qui est particulièrement utile lorsqu'aucune donnée RNAomique n'est disponible. MIDAS est capable d'aligner des ensembles de données hétérogènes et d'identifier des types de cellules, même de nouveaux types, lors du transfert de connaissances entre différents tissus.
L'analyse multi-omique unicellulaire continue de progresser
Tout comme nous pouvons voir le monde à travers un grain de sable, les scientifiques peuvent également voir le multivers, ou plus précisément, la « multi-omique », depuis l’intérieur d’une minuscule cellule.
Une gamme de techniques différentes est utilisée pour étudier le génome, le transcriptome, l’épigénome et d’autres caractéristiques des cellules individuelles, et bien que chaque technique soit informative en soi, leur analyse combinée – connue sous le nom de multi-omique – fournit une image plus complète.Actuellement, grâce à la multiomique unicellulaire, la biologie cellulaire et la recherche translationnelle ont réalisé des progrès significatifs, mais l’intégration et l’analyse des données restent des défis pour de nombreux scientifiques.
Sur cette base, en plus de l'équipe Ying Xiaomin et de l'équipe Bo Xiaochen mentionnées ci-dessus, d'autres équipes de recherche et entreprises suivent le mouvement, essayant d'explorer des moyens plus efficaces et plus simples de traitement des données.
Par exemple,Les méthodes analytiques telles que la plateforme monocellulaire Chromium de 10x Genomics continuent de se développer, permettant l'évaluation de multiples caractéristiques cellulaires dans différentes combinaisons.Y compris l'expression des gènes du transcriptome entier, l'expression des protéines et l'appariement complet TCR et séquençage BCR, spécificité antigénique et analyse de la chromatine ouverte. Parmi eux Ranger de cellule La solution utilise un ensemble de pipelines d'analyse gratuits et faciles à utiliser pour analyser les données unicellulaires de Chromium, qui peuvent traiter les données brutes et effectuer des alignements pour compter les gènes. De plus, Cell Ranger peut également être intégré à des plateformes d’analyse cloud pour surveiller, gérer et traiter les données.
Par exemple,Le 2 mai 2022, le groupe de recherche de Gao Ge à l'Université de Pékin/Laboratoire Changping a publié un article de recherche intitulé « Intégration de données monocellulaires multi-omiques et inférence réglementaire avec intégration liée au graphe » dans Nature Biotechnology.Une méthode d'apprentissage profond appelée GLUE basée sur une stratégie de couplage de graphes a été proposée, qui a permis pour la première fois une intégration précise non supervisée et une inférence réglementaire de millions de données multi-omiques unicellulaires.
Le développement continu de ces outils et logiciels bioinformatiques aidera les chercheurs à interpréter des ensembles de données multi-omiques complexes et à promouvoir le développement de la biologie cellulaire. Il est d’une grande importance pour révéler les fonctions et les mécanismes de régulation moléculaire des cellules et étudier l’apparition et le développement des maladies, et en fin de compte bénéficier aux populations.
Références :
1.https://www.chinagut.cn/articles/ss/02bc1e86e3734acebff57395d6e044a6
2.https://m.ebiotrade.com/newsf/2023-10/20231023151001602.htm
3.https://news.bioon.com/article/e49a810955a1.html
4.https://m.thepaper.cn/newsDetail_forward_26137031








