Command Palette
Search for a command to run...
Le Groupe De Recherche De Li Huashan Et Wang Biao À l'Université Sun Yat-sen a Développé Le Modèle d'apprentissage Automatique SEN Pour Prédire Les Propriétés Des Matériaux Avec Une Grande précision.

Sommaire en un coup d'œil:La compréhension de la symétrie globale des cristaux et l’analyse des informations équivariantes sont essentielles pour prédire les propriétés des matériaux, mais les algorithmes existants basés sur les réseaux convolutifs ne peuvent pas répondre pleinement à ces exigences. Pour résoudre ce problème, le groupe de recherche dirigé par Li Huashan et Wang Biao de l'Université Sun Yat-sen a développé un modèle d'apprentissage automatique appelé SEN, qui perçoit avec précision l'interaction entre la symétrie cristalline inhérente et les groupes de structures matérielles.
Mots-clés: Base de données MP d'apprentissage profond pour la prédiction des propriétés des matériaux
Auteur | Li Baozhu
Rédacteur | Sanyang
La symétrie cristalline joue un rôle clé dans l’étude des propriétés physiques des matériaux, la compréhension de la structure cristalline, la conception de nouveaux matériaux et la réalisation d’expériences telles que la diffraction des rayons X. La compréhension des symétries cristallines peut aider à simplifier les analyses, à mieux comprendre les propriétés des matériaux et à rendre les calculs de performance des matériaux plus efficaces. Plus important encore, la symétrie cristalline peut également affecter directement la distribution de charge du matériau, ses propriétés optiques, ses propriétés magnétiques et d’autres propriétés physiques.
Ces dernières années, l’apprentissage automatique basé sur des mécanismes statistiques a été largement utilisé. Du point de vue de l’apprentissage automatique, la symétrie cristalline peut être considérée comme l’invariance et l’équivariance des matériaux. Cependant, les algorithmes d’apprentissage automatique existants pour les matériaux cristallins basés sur des réseaux de graphes avancés ont du mal à coder l’invariance et l’équivariance complexes des matériaux.
De plus, bien que l'autoencodeur à capsule empilée (SCAE) puisse également extraire directement les caractéristiques de symétrie spatiale des données d'origine, le modèle de capsule traditionnel est toujours incapable d'analyser la relation entre la structure et les performances des systèmes de matériaux complexes.
Au vu des défis susmentionnés,Le groupe de recherche dirigé par Huashan Li et Biao Wang de l'Université Sun Yat-sen a développé un modèle d'apprentissage automatique appelé SEN (réseau d'équivariance amélioré par symétrie)., surmontant les faibles performances des algorithmes basés sur la convolution dans les groupes d'espaces à haute symétrie et obtenant des prédictions de propriétés matérielles de haute précision dans tous les groupes d'espaces. À l’heure actuelle, les résultats pertinents ont été publiés dans « Nature Communications ».

Les résultats connexes ont été publiés dans « Nature Communications »
Obtenez le papier :
https://www.nature.com/articles/s41467-023-40756-2
01 Ensemble de données : 6 027 matériaux cristallins dans la base de données MP
Les chercheurs ont extrait les caractéristiques des matériaux cristallins en se basant sur le concept d’environnement chimique et la méthode de représentation des modèles graphiques. Ils ont défini son environnement chimique par les atomes environnants et les liaisons dans le rayon de coupure de l'atome cible, et ont extrait le type d'atome, la connectivité atomique et la longueur de liaison autour de chaque atome du Materials Project, une base de données Python open source pour l'analyse des matériaux.
Il est rapporté que,Les ensembles de données utilisés pour prédire la bande interdite et l'énergie de formation dans cette étude proviennent de la base de données Materials Project, et les ensembles de données de bande interdite et d'énergie de formation contiennent respectivement 6 027 (divisés en ensemble d'entraînement, ensemble de validation et ensemble de test dans un rapport de 8:1:1) et 30 000 matériaux.Les deux ensembles de données comprennent 64 éléments, couvrant les éléments du tableau périodique à l'exception du groupe des gaz nobles, des lanthanides, des actinides et des éléments radioactifs.
Les chercheurs ont utilisé des calculs de théorie fonctionnelle de la densité (DFT) pour prédire la composition de 6 027 matériaux cristallins dans la base de données Materials Project et ont testé les performances du modèle SEN sur la base des conclusions prédites.
Les données de symétrie cristalline et d’environnement chimique utilisées dans cette étude sont disponibles dans la base de données Zenodo.
Visitez le lien:
https://doi.org/10.5281/zenodo.8142678
02 Architecture du modèle : formation unifiée de 3 modules
Comme le montre la figure ci-dessous,Le modèle SEN adopte une architecture d'apprentissage profond complexe, qui comprend des modules d'extraction de caractéristiques (FE), de perception de symétrie (SP) et de prédiction de propriétés (PP).
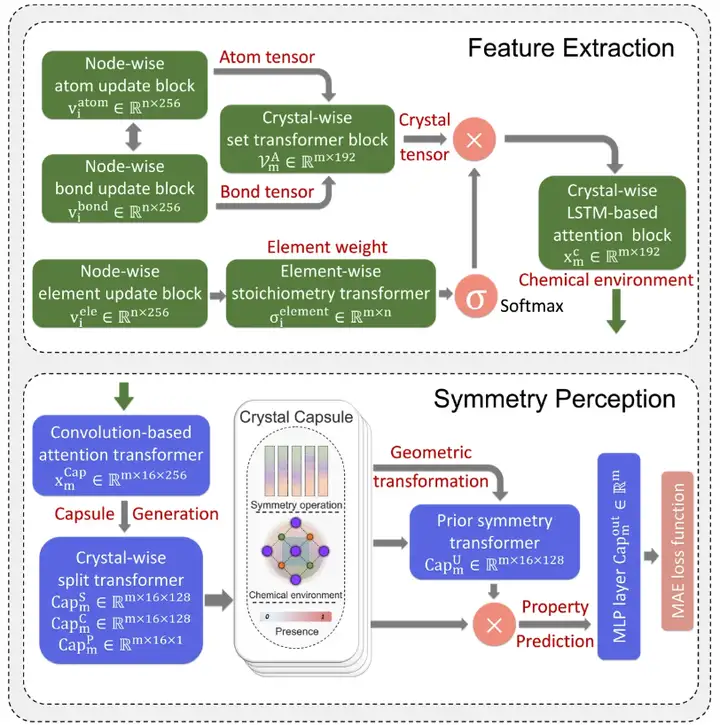
L'architecture SEN se compose de modules d'extraction de caractéristiques, de perception de symétrie et de prédiction d'attributs.
Dans cette étude, l’équipe de recherche a réussi à prédire avec précision les propriétés de plusieurs matériaux grâce à une formation unifiée de trois modules et a décrit l’interaction entre les atomes grâce au modèle SEN.
Tout d’abord, le module d’extraction de caractéristiques détecte les données d’entrée atomiques et de liaisons chimiques, qui incluent les informations sur les atomes N et les liaisons M dans l’unité d’origine du matériau cible. Enfin, grâce à un processus de criblage à haut débit, un ensemble de données sur les matériaux comprenant la stoechiométrie, la structure cristalline, les informations atomiques et les informations de liaison a été construit.
En utilisant l'ensemble de données matérielles comme seules données d'entrée pour le modèle SEN, les chercheurs ont calculé simultanément le vecteur d'environnement chimique atomique VmA et le vecteur de poids des éléments VmE sur la base des données structurelles et des données stoechiométriques.
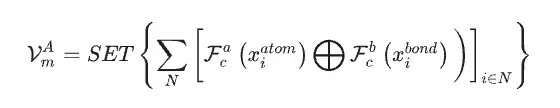
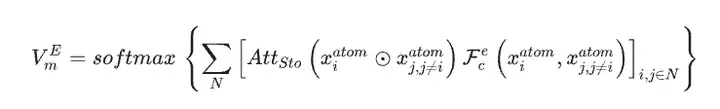
Après activation par le perceptron multicouche, le vecteur de poids de l'élément est converti en vecteur de probabilité de l'atome correspondant. Les chercheurs ont ensuite mis à jour toutes les corrélations au niveau atomique par le biais d'opérations élément par élément entre les vecteurs d'environnement chimique atomique et les vecteurs de poids des éléments, obtenant ainsi la matrice d'environnement chimique du matériau via la couche d'attention LSTM.
Deuxièmement, cette étude a appliqué de manière innovante le mécanisme de la capsule à la prédiction des propriétés des matériaux. Grâce au module de perception de symétrie conçu sur la base du mécanisme de capsule, l'environnement chimique du matériau a été converti en une capsule matérielle composée d'opérateurs de symétrie, d'un environnement chimique du matériau convolutif et de valeurs d'existence pour percevoir et préserver la symétrie cristalline. De plus, différents modèles de symétrie peuvent être généralisés aux capsules cristallines en effectuant des opérations de symétrie sur la matrice de l'environnement chimique du matériau.
Enfin, en termes de prédiction des propriétés, le modèle SEN prédit les propriétés du matériau cible via une fonction de mappage basée sur MLP.
Le modèle 03 SEN prédit les propriétés des matériaux avec une grande précision
Conclusion 1 : Le modèle SEN perçoit avec précision les informations d'interaction atomique
Pour vérifier l'efficacité du module d'extraction de caractéristiques, les chercheurs ont entraîné la capacité du SEN à prédire la bande interdite des matériaux cristallins jusqu'à ce que l'erreur absolue moyenne (MAE) soit inférieure à 0,15 eV, puis ont analysé les données intermédiaires de l'environnement chimique générées par le module d'extraction de caractéristiques.
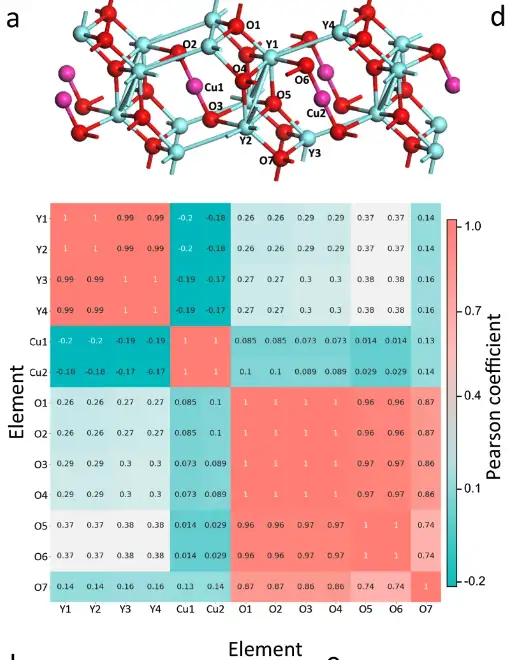
Analyse de corrélation de l'environnement chimique basée sur les atomes
Plus précisément, les chercheurs ont extrait la matrice de l’environnement chimique de chaque atome dans la cellule unitaire de Y4Cu2O7. Le coefficient de Pearson entre les matrices atomiques a été calculé, générant le graphique d'analyse de corrélation présenté ci-dessus. Les coefficients de Pearson entre les atomes du même groupe d'éléments sont beaucoup plus grands que ceux entre les atomes de différents groupes d'éléments, de sorte que les trois groupes d'éléments dans Y4Cu2O7 peuvent être clairement distingués.
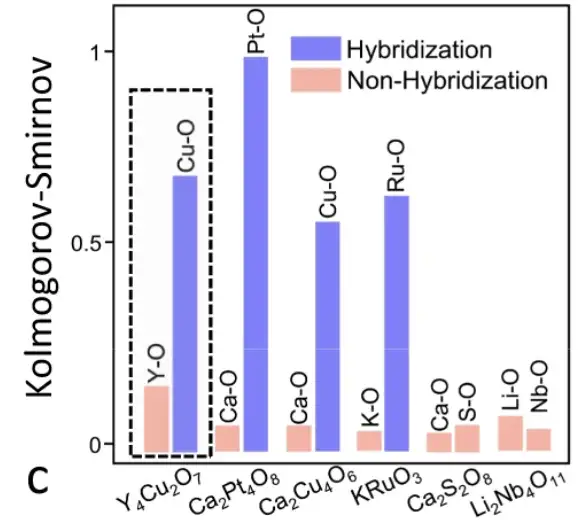
Les corrélations atomiques de six matériaux ont été apprises par le modèle SEN
Comme le montre la figure ci-dessus, le modèle SEN a appris et codé les informations d'interaction atomique et détecté avec succès le phénomène d'hybridation, ce qui est d'une grande importance pour la prédiction des propriétés électroniques.
Conclusion 2 : Les performances de prédiction du modèle SEN sont meilleures que celles de MegNet
Pour étudier la cartographie de l'environnement chimique aux propriétés des matériaux dans le modèle SEN, les chercheurs ont sélectionné cinq matériaux de la base de données MP - Be(6)Ni(2), Sr(4)Ge(2)S(8), Li(2)V(2)F(12), CsAsF(6) et BaB(2)F(8), avec des bandes interdites de 0 eV, 3,25 eV, 4,86 eV, 7,24 eV et 10,12 eV, respectivement.
On observe qu'il existe une forte corrélation entre la bande interdite et la PDF (fonction de densité de probabilité) de l'environnement chimique du matériau, c'est-à-dire que la PDF s'étale progressivement à mesure que la bande interdite augmente. La projection de l’ensemble des données de l’environnement chimique du matériau à la bande interdite est illustrée dans la figure ci-dessous. Les 6 027 matériaux cristallins sont répartis uniformément dans l'espace principal, tandis que le changement de bande interdite est continu et monotone dans tout l'espace.
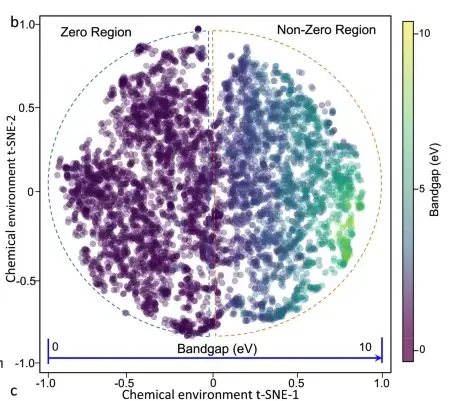
Graphique t-SNE 2D de 6027 matériaux. La couleur du cercle indique la valeur de la bande interdite.
Pour vérifier que les relations caractéristiques-attributs apprises par le modèle d'apprentissage automatique sont cohérentes avec les principes physiques de base, les chercheurs ont généré une carte t-SNE 2D de l'environnement chimique du matériau Ca-OX et ont étudié diverses caractéristiques du matériau (composition, groupe de points, polarisation de spin, etc.). Ils ont finalement découvert que la bande interdite du matériau dépend de caractéristiques complexes du matériau et ne peut pas être simplement prédite par un facteur clé.
Néanmoins, le modèle SEN permet d’obtenir des améliorations significatives dans la prédiction de la bande interdite.Le modèle SEN atteint une erreur quadratique moyenne (MAE) de 0,25 eV lors de la prédiction de la bande interdite des matériaux dans l'ensemble de données de test, ce qui constitue une amélioration significative par rapport à la MAE obtenue par les modèles avec les modules MLP, DenseNet, TFN, SE(3) et EGNN sur l'ensemble de données de test.
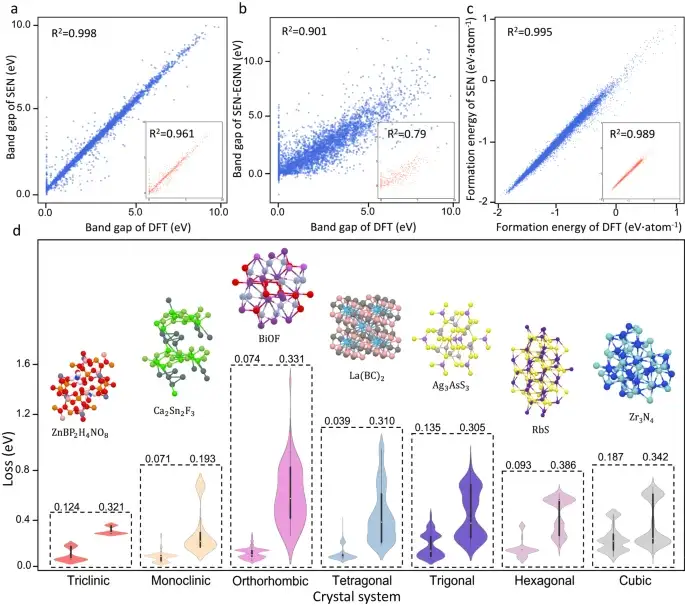
Prédiction des propriétés des matériaux cristallins avec différentes symétries
Comme le montre la figure d ci-dessus, les chercheurs ont comparé la qualité de prédiction du modèle SEN et du modèle MegNet21 (modèle de réseau de matériaux général) pour différents systèmes cristallins, révélant en outre l'impact significatif de la perception de la symétrie sur la prédiction des propriétés des matériaux.À partir du diagramme de distribution des erreurs, on peut voir que les performances de prédiction du modèle SEN sont meilleures que celles de MegNet dans tous les systèmes cristallins.
De plus, le modèle SEN réduit considérablement la dimension caractéristique effective en détectant la symétrie complète du cristal. Ce processus de nettoyage des fonctionnalités atténue le problème de surapprentissage et renforce le mappage des fonctionnalités matérielles aux propriétés.
L'article montre queLes erreurs absolues moyennes de la bande interdite et de l'énergie de formation prédites par le modèle SEN sont respectivement environ 22,9% et 38,3% inférieures à celles des modèles d'apprentissage automatique courants.
04 L'IA favorise la transformation et le développement de l'industrie des matériaux
Pendant longtemps, la conception, la recherche et le développement de nouveaux matériaux et la réforme des propriétés des matériaux ont été l'un des moteurs du progrès scientifique et technologique, jouant un rôle important dans de nombreux domaines tels que l'électronique, l'énergie, les soins médicaux, l'aérospatiale, etc. Cependant, le processus traditionnel de recherche et de développement des matériaux nécessite souvent un grand nombre d'expériences pour corriger en permanence les performances et améliorer la faisabilité. Ce processus est long et nécessite d’énormes ressources humaines et financières.
Avec l’application accélérée de l’IA, l’IA pour la science a reçu de plus en plus d’attention, et sa combinaison avec les matériaux est devenue une nouvelle direction d’exploration pour de plus en plus de chercheurs et d’entreprises. D’une part, l’IA peut analyser de grandes quantités de données et effectuer des prédictions de simulation, accélérant ainsi la découverte de nouveaux matériaux et optimisant leurs performances. D’autre part, la science des matériaux est également devenue un point d’appui important pour les technologies clés de l’IA telles que l’apprentissage automatique, le traitement du langage naturel et le calcul haute performance.
On peut dire que l’IA modifie tranquillement la conception et l’application de nouveaux matériaux. À l’avenir, avec l’itération continue de modèles d’IA plus puissants et la mise à jour et l’expansion des bases de données de matériaux dans le cadre du partage de données, l’IA est vouée à favoriser davantage la naissance de nouveaux matériaux.








