Command Palette
Search for a command to run...
Stratégie De Sélection Des Caractéristiques : Trouver De Nouvelles Façons De Détecter Les Biomarqueurs Du Cancer Du Sein

Contenu en un coup d'œil :Le microARN (microARN) est une classe de transcrits d'ARN non codants courts et monocaténaires. Ces molécules présentent une croissance incontrôlée dans une variété de tumeurs malignes et ont donc été identifiées par de nombreuses études ces dernières années comme des biomarqueurs fiables pour le diagnostic du cancer. Parmi les différentes analyses pathologiques, l’analyse d’expression différentielle est souvent considérée comme une méthode efficace pour détecter des biomarqueurs clés. Des chercheurs de l'Université de Naples Federico II en Italie ont suggéré qu'une stratégie de sélection de caractéristiques basée sur l'apprentissage automatique peut être plus efficace pour la détection, et ont recommandé que les 20 microARN qu'ils ont découverts soient utilisés comme biomarqueurs diagnostiques du cancer du sein.
Mots-clés:Sélection de caractéristiques microARN Cancer du sein
Cet article a été publié pour la première fois sur la plateforme publique HyperAI WeChat~
Selon l'édition 2022 des « Lignes directrices pour le traitement du cancer du sein » publiées par la Commission nationale de la santé,Le cancer du sein est l’une des tumeurs malignes les plus courantes chez les femmes et son taux d’incidence se classe au premier rang parmi les tumeurs malignes chez les femmes.Selon les statistiques de l’Organisation mondiale de la santé, en 2020, 2,3 millions de femmes dans le monde ont reçu un diagnostic de cancer du sein. Avec l’amélioration continue des méthodes de traitement,Le taux de survie à cinq ans pour le cancer du sein précoce peut atteindre 90% ou même plus. Par conséquent, un diagnostic précis du cancer du sein à un stade précoce est particulièrement important.
En plus de jouer de nombreux rôles clés en biologie, les changements dans l’expression des microARN sont également associés à une variété de cancers, ils peuvent donc être utilisés comme un biomarqueur diagnostique putatif fiable. Des chercheurs de l'Université de Naples Federico II en Italie ont utilisé l'apprentissage automatique pourEn utilisant une stratégie de sélection de caractéristiques et en analysant la stabilité et les performances de classification de trois méthodes,Un panel de biomarqueurs diagnostiques spécifiques du cancer du sein a été obtenu et des gènes clés putatifs dans le développement et la progression du cancer du sein ont été découverts.
Actuellement, ce résultat de recherche a été publié dans les actes de la 18e conférence sur les méthodes d'intelligence computationnelle en bioinformatique et biostatistique (CIBB 2023) sous le titre « Une stratégie de sélection de caractéristiques robuste détecte un panel de microARN comme biomarqueurs diagnostiques putatifs dans le cancer du sein ».

Les résultats de la recherche ont été publiés dans CIBB 2023
Adresse du document :
https://www.researchgate.net/publication/372083934
Aperçu de l'expérience
Dans cette étude, les chercheurs ont découvert qu'à l'aide de trois méthodes de sélection de caractéristiques (taux de gain, forêt aléatoire et élimination récursive des caractéristiques par machine à vecteurs de support), les combinaisons moléculaires diagnostiques peuvent être extraites plus efficacement. Ils ont révélé un panel de 20 microARN, parmi lesquels hsa-mir-337, hsa-mir-378c et hsa-mir-483 n'ont pas reçu une attention généralisée dans la communauté médicale parmi les biomarqueurs diagnostiques actuels du cancer du sein. Cette méthode permet de distinguer les échantillons sains des échantillons tumoraux. Par rapport à la méthode d'expression différentielle couramment utilisée, ses performances de classification sont meilleures et il est plus facile d'identifier les caractéristiques qui sont facilement sous-estimées ou même ignorées.
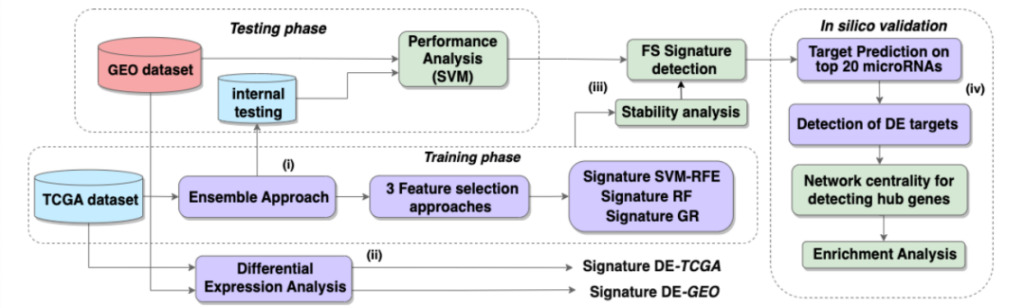
Figure 1 : Aperçu du pipeline
Le flux de travail comprend 4 étapes clés :
(je) Calculs d'ensemble-FS sur le sous-ensemble d'entraînement TCGA
(ii) Analyse d'expression différentielle des ensembles de données TCGA/GEO
(iii) Comparer les performances de classification de l'analyse d'expression différentielle et les résultats de sélection de caractéristiques, et évaluer la stabilité des méthodes de sélection de caractéristiques
(iv) La simulation informatique a été utilisée pour vérifier les 20 premiers microARN de la signature sélectionnée et pour détecter les cibles des gènes centraux.
Détails de l'expérience
Ensemble de données
Les sources de données expérimentales comprennent deux canaux :Le projet TCGA-BRCA sur le site officiel du GDC américain et le référentiel de données Gene Expression Omnibus (GEO) (GSE97811).
L'équipe expérimentale a collecté un total de 1 881 données microRNA-Seq du projet GDC TCGA-BRCA et les a divisées en ensembles d'entraînement et de test dans un rapport de 8:2. Les données ont été associées à 300 échantillons de tumeurs primaires solides (T) et 101 échantillons de tumeurs adjacentes normales (NAT), à la fois de tissu mammaire canalaire et lobulaire. Avant d’appliquer la sélection de fonctionnalités,Ces données ont été soumises à une normalisation stabilisatrice de la variance.
Dans le même temps, les chercheurs ont téléchargé un ensemble de données de puces à ADN contenant 2 565 microARN de la base de données GEO (GSE97811).Il est utilisé comme ensemble de validation pour cette expérience.L'ensemble de données comprend 16 échantillons normaux et 45 échantillons tumoraux, puis l'imputation des données est effectuée.
Étant donné que les données GEO (l'ensemble de validation de cette expérience) impliquent l'expression de microARN matures et que les données TCGA (l'ensemble d'entraînement et l'ensemble de test de cette expérience) contiennent la forme précurseur, afin d'unifier les données, les chercheurs ont uniquement sélectionné des microARN matures alternatifs avec des valeurs de comptage moyennes supérieures à leurs brins opposés dans les échantillons de données GEO ; dans le même temps, les noms des microARN ont été convertis en noms de formes précurseurs correspondantes.
Après ce processus,La dimensionnalité des données GEO (ensemble de validation) a été réduite à 1 361 microARN, et les données TCGA RNA-Seq correspondantes ont également été collectées, comprenant un total de 20 404 gènes.
1. Méthode de sélection des fonctionnalités et application du programme Ensemble
Les chercheurs ont sélectionné trois méthodes de sélection de caractéristiques à comparer avec l’analyse d’expression différentielle.Ils sont Rapport de gain, forêt aléatoire et SVM-RFE (Support Vector Machine Recursive Feature Elimination).Trois méthodes ont été appliquées à 500 sous-ensembles de données TCGA d'expression de microARN-Seq pour identifier un panel de caractéristiques robustes capable de distinguer les échantillons normaux des échantillons tumoraux. Dans les résultats observés, les données ont été divisées en un ensemble d'entraînement et un ensemble de test dans un rapport de 8:2, puis les données ont été rééchantillonnées et amorcées pour les rendre conformes à la procédure d'ensemble de perturbation des données. Chaque calcul renvoie 500 vecteurs microARN triés par ordre décroissant de « score d'importance ».
|Remarques :Le score d’importance représente l’influence de chaque caractéristique dans la classification calculée par l’algorithme.
Plus le score d’importance est élevé, plus le rang attribué à la fonctionnalité est bas.Les chercheurs ont ensuite utilisé une procédure d’agrégation pour dériver une signature consensuelle pour chaque méthode de sélection de caractéristiques et ont finalement retenu les 200 principales caractéristiques pour chaque groupe de microARN.
2. Test de stabilité
L'indice de Kuncheva (KI) et le pourcentage de gènes/caractéristiques qui se chevauchent (POG) ont été utilisés pour évaluer la cohérence des méthodes de sélection des caractéristiques, et la statistique Stot (mesure par paire de KI) a été utilisée pour déterminer la stabilité entre toutes les méthodes.Ces statistiques sont calculées à mesure que la longueur de la signature augmente.Le nombre de fonctionnalités commence à 2 et se termine à 200, et chaque recalcul augmente de 2 unités.

Formule statistique Stot
3. Analyse d'expression différentielle et signature DE
Une analyse d'expression différentielle a été réalisée sur des ensembles de données TCGA (y compris microRNA-Seq et RNA-Seq), en commençant par les comptages bruts, en utilisant le test exact, puis en conservant les caractéristiques DE avec FDR <= 0,01 et un seuil Log2FC de |0,5|.Pour obtenir la signature du DE-microARN, les valeurs Log2FC ont été converties en valeurs absolues, et les microARN (les 200 premières caractéristiques ont été conservées) ont été triés dans l'ordre décroissant d'abs (Log2FC).
L'ensemble de validation GEO a été exprimé de manière différentielle à l'aide de Limma, et les paramètres et procédures d'obtention des signatures DE dans cet ensemble de données étaient cohérents avec ceux de l'ensemble de données TCGA.
4. Analyse des performances de classification
Pour déterminer la capacité de chaque signature à distinguer les personnes en bonne santé des patients atteints de cancer,Les chercheurs ont mené une analyse prédictive sur quatre signatures (y compris des panels de sélection de caractéristiques et des panels d'expression différentielle) sur le sous-ensemble de test (TCGA) et l'ensemble de validation (GEO).
Enfin, la précision moyenne (ACC), la statistique K (KK) et le coefficient de corrélation de Matthews (MCC) sont calculés pour chaque pli et plusieurs longueurs de chaque signature.
5. Détection de la cible de signature microARN SVM-RFE
Pour identifier les cibles génétiques potentielles du microARN,Les chercheurs ont effectué les opérations suivantes :
1. Les 20 principaux microARN SVM-RFE ont été classés selon qu'ils étaient régulés à la hausse ou à la baisse dans les échantillons tumoraux.
2. Une analyse d'expression différentielle a été réalisée sur les données RNA-Seq pour détecter les gènes différentiellement exprimés (FDR <= 0,05).
3. L'analyse de corrélation de Spearman a été appliquée pour comparer l'expression des microARN avec les gènes différentiellement exprimés, et seuls les gènes up qui étaient négativement corrélés avec les microARN down et les gènes down qui étaient négativement corrélés avec les microARN up ont été conservés (rho <= -0,5).
4. Toutes les cibles génétiques microARN validées ont été collectées et seules celles qui présentaient également une corrélation DE ont été conservées.
6. Centralité du réseau et identification des gènes pivots
Matrice de corrélation (Spearman) de gènes dérégulés sélectionnés,Et utilisez-le pour construire un réseau de gènes à structure graphique :Les gènes hub avec un score de centralité hub de Kleinberg > 75, rho > 0,8 ou rho < -0,6 ont été conservés. Une analyse d'enrichissement génétique (ORA) a été réalisée sur les gènes centraux pour explorer les voies les plus enrichies de la base de données REACTOME. Le seuil de pValue ajusté par FDR a été fixé à 0,005.
Résultats expérimentaux
L'expérience a montré qu'après avoir appliqué les trois méthodes de sélection de caractéristiques, 500 signatures de microARN ont été renvoyées par ordre décroissant d'importance, et trois panels de consensus ont été obtenus après agrégation. Il est à noter que les trois principaux microARN (hsa-mir-139, hsa-mir-96 et hsa-mir-145) sont apparus dans tous les panels, démontrant l’importance de ces molécules pour distinguer les échantillons tumoraux des échantillons sains.
Conclusion 1 : SVM-RFE présente la plus grande stabilité
D'après le calcul de KI et POG sur le panel de consensus,La méthode SVM-RFE est la plus stable et est la plus importante lorsque la longueur de la signature atteint 20 caractéristiques. De même, les résultats de l’indice Stot montrent également que la méthode SVE-RFE présente la plus grande stabilité.
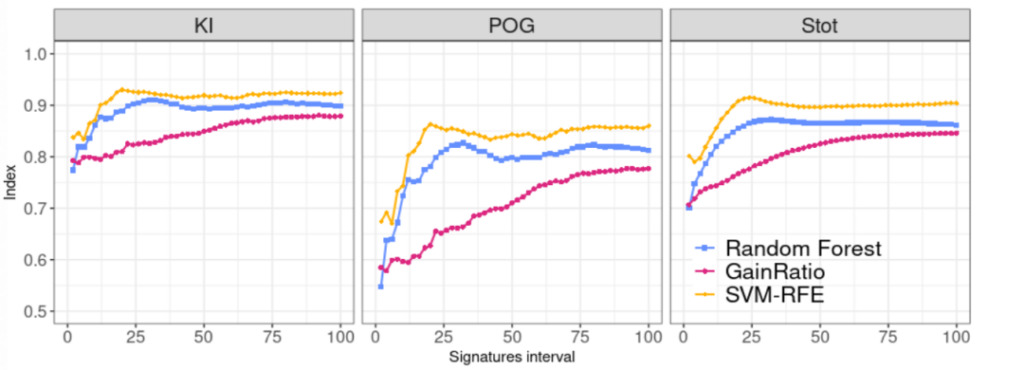
Figure 2 : Comparaison de l'indice de stabilité de trois méthodes de sélection de caractéristiques
bleu:Forêt aléatoire
rose:Rapport de gain
jaune:SVM-RFE (Suppression des caractéristiques récursives de la machine à vecteurs de support)
|Conclusion 2 : La signature SVM-RFE est plus performante que la signature d'expression différentielle en matière de classification
Après l'analyse des performances de classification de tous les panels individuels, l'ensemble de test (TCGA) et l'ensemble de validation (GEO) ont montré que la signature obtenue par SVM-RFE avait la capacité prédictive la plus élevée.
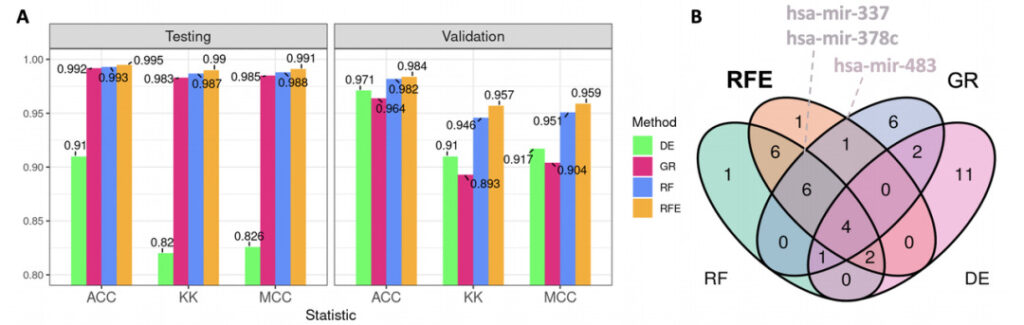
Figure 3 : Performances de classification des 20 principaux microARN et diagramme de Venn
UN:Le graphique à barres montre la statistique moyenne calculée sur le sous-ensemble de test et l'ensemble de données GEO de validation externe.
ACC:Précision
KK:Statistique K
MCC :Coefficient de corrélation de Matthews
vert:DE (analyse d'expression différentielle, la méthode de contrôle dans cette expérience)
rose:GR (rapport de gain)
bleu:RF (forêt aléatoire)
couleur orange:RFE (SVM-RFE, élimination des caractéristiques récursives de la machine à vecteurs de support)
B:Figure 4 : Diagramme de Venn des 20 premiers microARN pour chaque signature, avec quelques microARN intéressants parmi les 20 premiers du panel SVM-RFE étiquetés : hsa-mir-337, hsa-mir-378c et hsa-mir-483. Ces trois micro-ARN sont apparus dans les trois méthodes de sélection de caractéristiques, mais leur fiabilité en tant que preuve diagnostique n'a pas encore été entièrement déterminée dans les études actuelles sur le cancer du sein.
|Conclusion 3 : L’analyse de réseau révèle des gènes potentiellement clés dans l’évolution des maladies
Des expériences ont montré que CDC25, TPX2 et KIF18B sont fortement exprimés dans les cellules souches de différents types de cancer et de patients atteints d'un cancer du sein triple négatif, et que la régulation négative de TGFBR2 est associée à la progression du cancer.
MicroARN : un autre candidat idéal pour le dépistage précoce du cancer du sein
Les méthodes traditionnelles de dépistage du cancer du sein reposent encore sur l’imagerie radiographique et la biopsie tissulaire, qui ne permettent pas d’établir une compréhension plus approfondie et plus complète de l’ensemble du génome du cancer. Cette méthode est non seulement très invasive, coûteuse et sujette à des effets secondaires, mais elle donne également souvent des résultats faussement positifs ou faussement négatifs. Il est difficile d’améliorer la précision du dépistage précoce du cancer du sein et l’expérience des patientes.Il est toujours nécessaire de développer de nouvelles stratégies pour faire face au fardeau du cancer du sein.
Depuis sa première découverte en 1993, le micorRNA a continué d’approfondir notre compréhension du cancer et a montré un grand potentiel en tant que biomarqueur fiable pour le diagnostic du cancer du sein.
Le microARN est un petit ARN non codant d'une longueur d'environ 19 à 25 nt, qui peut réguler une variété de gènes cibles.Impliqué dans la régulation d'une variété de processus biologiques et pathologiques.En incluant la formation et le développement du cancer, on s'attend à ce qu'il compense les limites de l'imagerie radiographique et de la biopsie tissulaire actuelles en tant que méthodes de diagnostic courantes pour le dépistage du cancer du sein dans la pratique clinique.
Cependant, les applications cliniques matures du microARN n’ont pas encore été entièrement développées et un système d’évaluation de la sécurité pour l’utilisation du microARN n’a pas encore été établi.Il faudra probablement un certain temps avant que le microARN devienne la base diagnostique principale du cancer.
Articles de référence :
[1]https://www.who.int/zh/news-room/fact-sheets/detail/breast-cancer
[2]https://guide.medlive.cn/guideline/25596
[3]https://www.abcam.cn/kits/micrornas-as-biomarkers-in-cancer-1
[4]https://caivd-org.cn/webfile/file/20220508/20220508153691029102.pdf
[5]https://www.sohu.com/a/318088245_100120288
Cet article a été publié pour la première fois sur la plateforme publique HyperAI WeChat~








