Command Palette
Search for a command to run...
Le titre est vide. Veuillez fournir le titre à traduire.
Le titre est vide. Veuillez fournir le titre à traduire.
CNN pour la détection du cancer de la peau
Résumé
Please provide the title and abstract you would like me to translate into French.
One-sentence Summary
Addressing the tissue-of-origin bias that can confound prior cancer marker studies, this study developed three convolutional neural network architectures (1D-CNN, 2D-Vanilla-CNN, and 2D-Hybrid-CNN) to classify unstructured gene expression data from 10,340 TCGA tumor and 731 matched normal samples into 33 cancer types and normal tissues with 93.9-95.0% accuracy, while guided saliency analysis of the 1D-CNN model identified 2,090 concordant cancer markers.
Key Contributions
- Three convolutional neural network architectures (1D-CNN, 2D-Vanilla-CNN, and 2D-Hybrid-CNN) are developed to classify unstructured gene expression inputs into 33 cancer types or normal tissue categories.
- A guided saliency interpretation technique applied to the 1D-CNN model identifies 2,090 cancer marker genes that exhibit differential expression patterns concordant with their respective tumor types.
- Systematic evaluation on a combined TCGA dataset of 10,340 tumor and 731 matched normal samples demonstrates classification accuracies of 93.9% to 95.0% across 34 classes while mitigating tissue-of-origin bias.
Introduction
The authors leverage deep learning to address the critical need for accurate cancer classification and biomarker discovery from high-dimensional gene expression data. While previous machine learning approaches have achieved high diagnostic accuracy, they typically overlook the confounding influence of tissue-of-origin, which obscures the identification of true cancer-specific markers and limits functional interpretation. Additionally, prior studies lack systematic comparisons of how different neural network architectures impact predictive performance. To overcome these limitations, the authors design lightweight convolutional neural networks that simultaneously distinguish cancerous from normal tissue and classify specific tumor types. They also introduce a novel interpretation framework that isolates tissue-specific effects, enabling reliable extraction of biologically meaningful biomarkers for future clinical validation.
Dataset

-
Dataset composition and sources: The authors draw from a self-contained dataset provided directly within the article, primarily sourced from The Cancer Genome Atlas (TCGA). The collection encompasses a broad spectrum of malignancies, including adrenocortical cancer, bladder urothelial carcinoma, breast invasive carcinoma, glioblastoma multiforme, lung adenocarcinoma, and several other solid and hematologic tumors.
-
Key details for each subset: The provided excerpts do not specify exact subset sizes, filtering thresholds, or class distribution breakdowns. The authors simply note that the complete supporting dataset is embedded within the publication itself.
-
How the paper uses the data: The dataset serves as the foundational resource for the study's deep learning and convolutional neural network analyses. The authors apply it directly to generate the article's conclusions without relying on external data repositories or third-party splits.
-
Processing and metadata construction: No explicit cropping strategies, normalization pipelines, or metadata construction steps are outlined in the provided sections. The authors confirm that all data collection, analysis, and interpretation were conducted independently of the listed funding agencies, ensuring unbiased processing throughout the workflow.
Method
The authors leverage a suite of three distinct convolutional neural network (CNN) architectures designed to address the unique challenges of predicting cancer types from high-dimensional gene expression data. The overarching framework is structured around the premise that gene expression data is inherently unstructured, and thus, conventional CNN designs must be adapted to extract meaningful patterns without relying on predefined gene ordering. The models are built with a focus on simplicity and robustness, particularly given the limited sample sizes typical in genomic studies, which makes overfitting a critical concern.
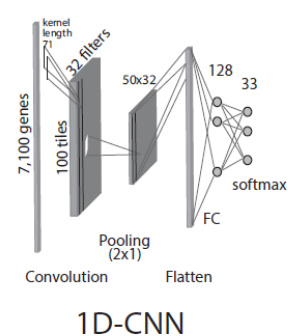
The first model, referred to as the 1D-CNN, treats the gene expression profile as a one-dimensional vector. As shown in the figure below, the input vector of 7,100 genes is processed by a single 1D convolutional layer with 32 filters, each with a kernel length matching the stride to capture global feature patterns rather than local dependencies. This design choice avoids the assumption of sequential correlation between adjacent gene expressions. The output of the convolution is passed through a max-pooling layer with a 2x1 window, followed by a flattening operation and a fully connected (FC) layer with 128 neurons, culminating in a softmax prediction layer for 33 cancer types and one normal class.
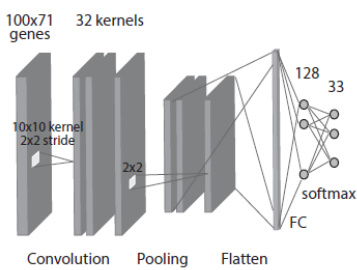
The second model, termed the 2D-Vanilla-CNN, transforms the gene expression vector into a 2D matrix, specifically a 100x71 grid, to conform to standard CNN input formats. The input is then processed by a 2D convolutional layer using 32 kernels of size 10x10 with a stride of 2x2, followed by a max-pooling layer with a 2x2 window. The output is flattened and fed into the same FC and softmax layers as the 1D-CNN. This architecture follows a conventional computer vision approach, where the 2D structure is treated as an image-like representation, though no specific biological ordering is imposed on the gene arrangement.
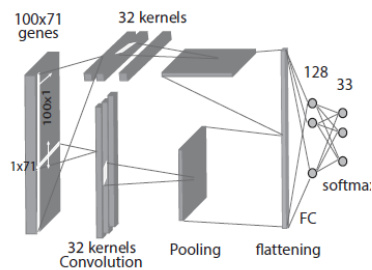
The third model, the 2D-Hybrid-CNN, combines the 2D input structure with 1D convolutional operations inspired by the parallel tower design in ResNet. As illustrated in the figure below, this model applies two separate 1D convolutional kernels—one sliding vertically along the rows and the other horizontally along the columns—over the 100x71 input matrix. The outputs from these two parallel convolutions are concatenated and then processed through a max-pooling layer, a flattening step, and the same FC and softmax layers. This hybrid approach aims to capture both row-wise and column-wise features efficiently while maintaining a simpler parameter structure compared to a full 2D CNN.
Additionally, the authors implement a 2D-3Layer-CNN model from a prior study to serve as a baseline for comparison. This model consists of three cascaded CNN modules, each incorporating batch normalization, activation functions, and max-pooling, followed by two FC layers and a softmax layer. The design of all models prioritizes computational efficiency and generalization, particularly by minimizing the number of trainable parameters, which is crucial given the constraints of genomic data availability.
Experiment
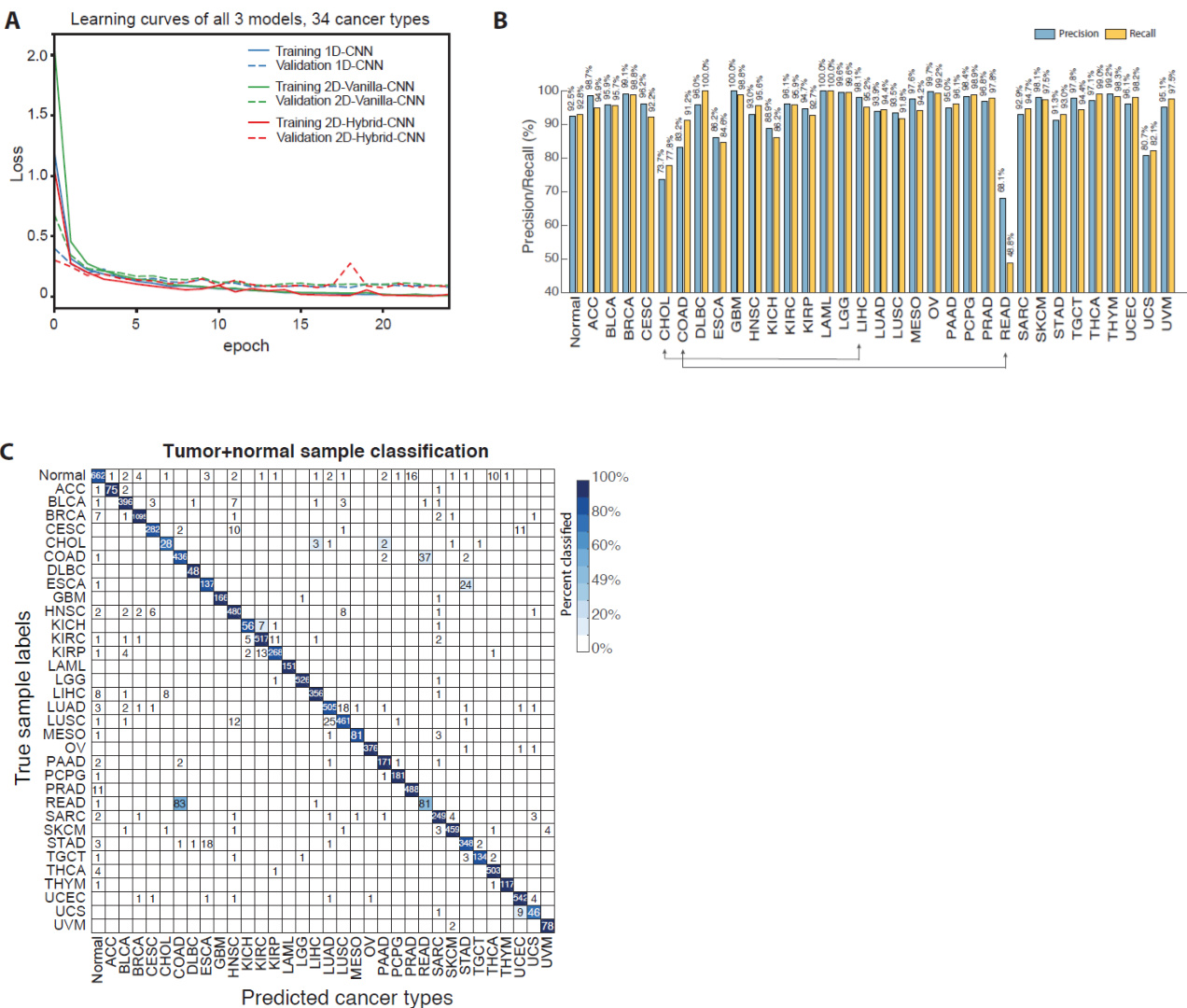
Three convolutional neural network architectures were trained on large-scale gene expression datasets to evaluate their capacity for cancer type and subtype classification. The experimental setup validated that these models achieve robust convergence and high accuracy while inherently capturing tissue-of-origin signatures, which were successfully decoupled by incorporating normal tissue controls to isolate cancer-specific signals. Interpretability analyses further demonstrated that the networks effectively identify biologically relevant marker genes with distinct expression profiles, aligning with known oncological pathways and revealing the inherent challenges in distinguishing histologically similar malignancies. Collectively, these findings confirm that deep learning approaches deliver reliable diagnostic predictions while providing transparent, mechanistically grounded insights into cancer genomics.
The authors evaluate three CNN models for cancer type prediction using gene expression data, comparing performance when trained on tumor samples only versus with additional normal samples. Results show that all models converge quickly and maintain low loss with minimal overfitting, while including normal samples slightly reduces overall accuracy but improves robustness. The 1D-CNN model achieves high classification accuracy and reveals patterns of misclassification, particularly among closely related cancer types. All three models converge to low loss within 10 epochs with no signs of overfitting when trained on tumor samples only. Including normal samples in training reduces overall accuracy slightly but improves model robustness and reduces bias toward tissue of origin. The 1D-CNN model shows high precision and recall for most cancer types, with notable misclassification among closely related types such as kidney and lung cancers.
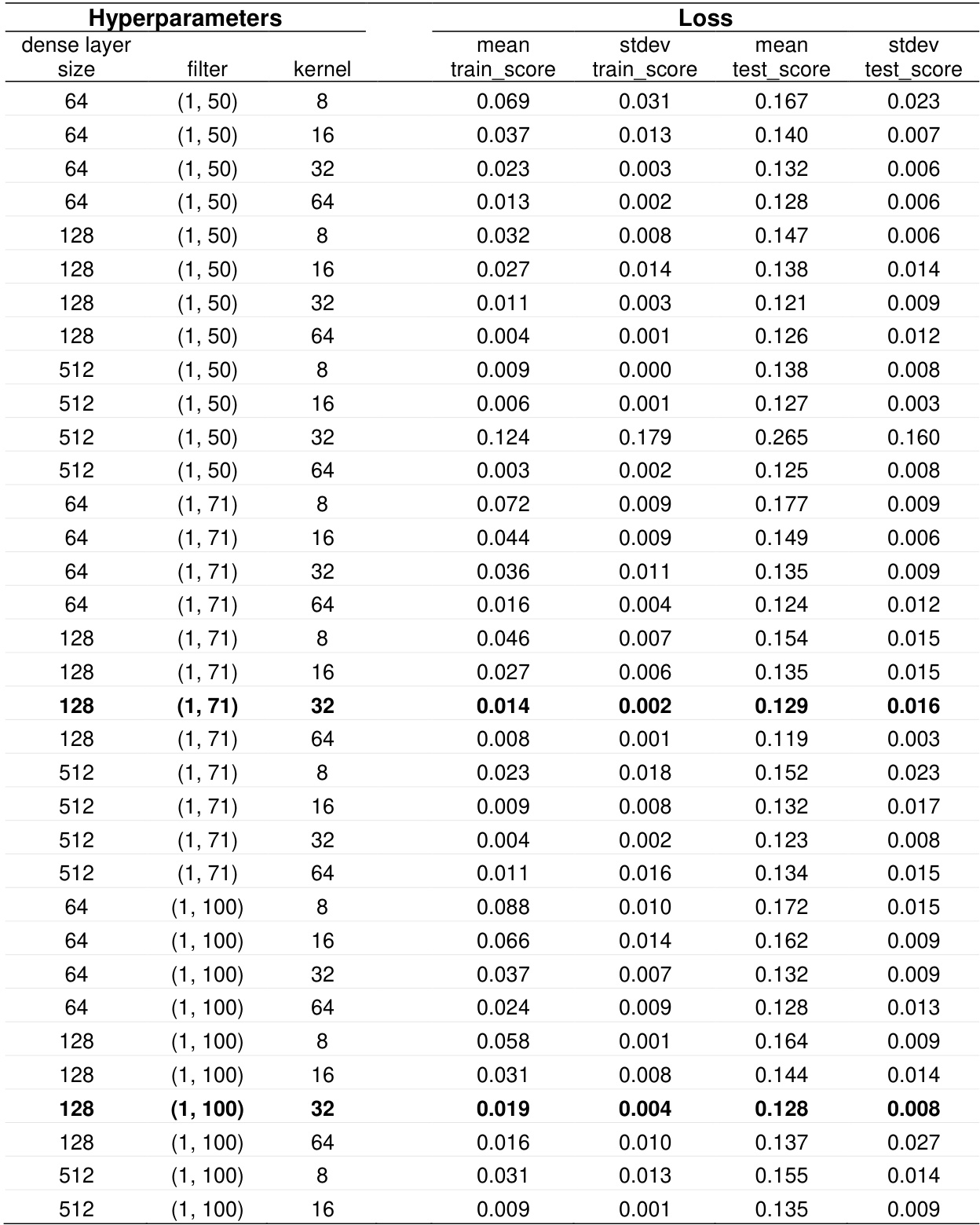
The the the table presents a grid search of hyperparameter combinations for a 1D-CNN model, showing how different configurations affect training and testing performance. The results indicate that the model's performance varies significantly with hyperparameter settings, particularly in test score and loss, with some configurations yielding lower test loss and more stable performance. Different hyperparameter settings lead to significant variations in model performance metrics like test score and loss. Configurations with a dense layer size of 128 and a kernel size of 32 show lower test loss and higher stability. The model performance is sensitive to the combination of dense layer size and kernel size, with some settings leading to higher test scores and lower loss values.
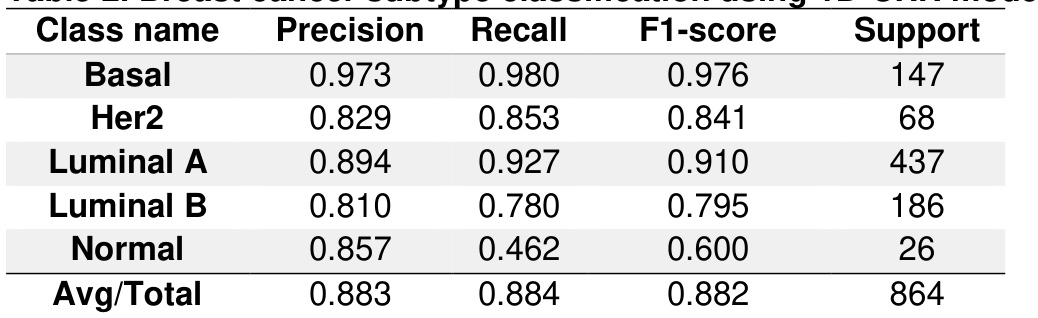
The authors trained a 1D-CNN model to predict breast cancer subtypes, including basal, HER2, luminal A, and luminal B, along with normal breast tissue. The model achieved high precision and recall for most subtypes, with particularly strong performance for the basal and luminal A classes, while the normal class showed lower recall. The overall average precision across all subtypes was high, indicating effective classification. The model achieved high precision and recall for basal and luminal A subtypes, with the highest precision observed for basal. The normal class had significantly lower recall compared to other subtypes, indicating weaker classification performance. The overall average precision across all breast cancer subtypes was high, reflecting strong model performance.
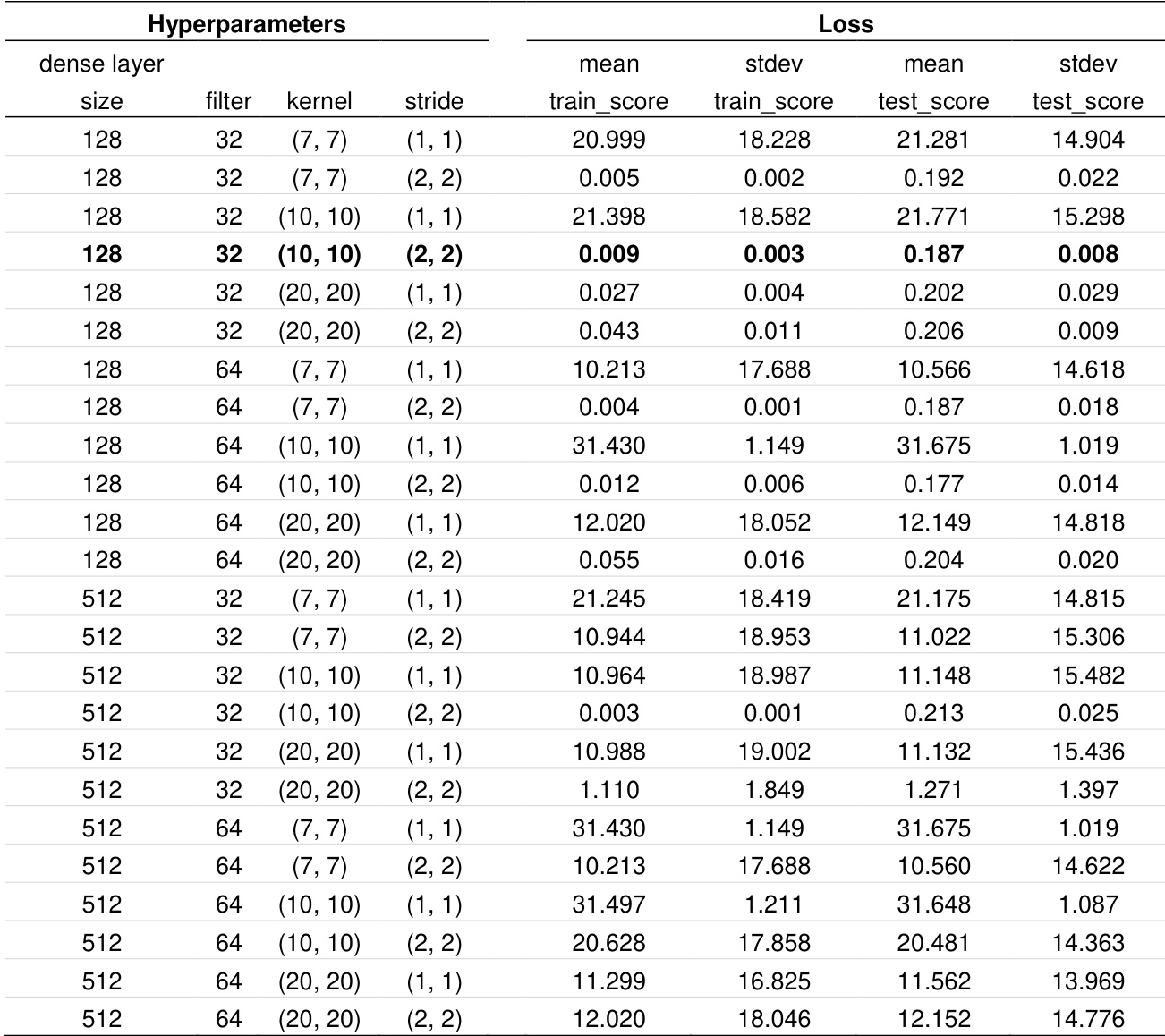
The the the table presents a grid search of hyperparameter combinations for a 1D-CNN model, evaluating different dense layer sizes, filter counts, kernel dimensions, and strides. The results show that the configuration with a dense layer size of 128, 32 filters, a (10, 10) kernel, and a (2, 2) stride achieved the lowest training and test loss, indicating optimal performance among the tested settings. The optimal hyperparameter configuration for the 1D-CNN model resulted in the lowest training and test loss. Increasing the dense layer size from 128 to 512 led to higher training and test loss for most parameter combinations. The (10, 10) kernel with a (2, 2) stride combination consistently produced lower test loss compared to other kernel and stride combinations.
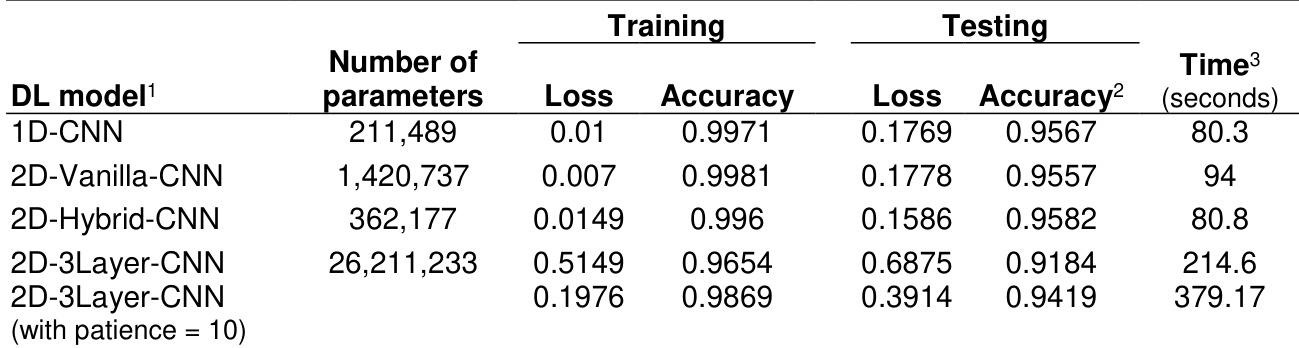
The the the table presents performance metrics for four deep learning models, including training and testing loss and accuracy, along with training time. The 2D-Vanilla-CNN model exhibits the highest training accuracy and the lowest training loss, while the 1D-CNN and 2D-Hybrid-CNN models show similar testing performance. The 2D-3Layer-CNN model has the highest training time and a notably lower testing accuracy compared to the other models. The 2D-Vanilla-CNN model achieves the highest training accuracy and the lowest training loss among the models. The 1D-CNN and 2D-Hybrid-CNN models show comparable testing accuracy, with the 2D-Hybrid-CNN slightly higher. The 2D-3Layer-CNN model has the longest training time and the lowest testing accuracy.
The experiments evaluate multiple CNN architectures on gene expression data to classify cancer types and subtypes, comparing models trained exclusively on tumor samples against those augmented with normal tissue while systematically optimizing network hyperparameters. Results demonstrate that all architectures converge rapidly with stable training, and incorporating normal samples slightly reduces peak accuracy but significantly enhances robustness and mitigates tissue-origin bias. The 1D-CNN consistently demonstrates strong generalization across diverse categories, though it occasionally confuses biologically similar subtypes, while deeper 2D configurations tend to require longer training without improving generalization. Ultimately, the findings validate that moderate network depths paired with balanced training data yield the most reliable and efficient genomic classifiers.