Command Palette
Search for a command to run...
Machine Learning Force Field Training with VASP
Abstract
One-sentence Summary
The authors propose a universal multiscale higher-order equivariant machine learning force field integrated with active learning and a bond length stretching method, which achieves the highest predictive accuracy and magnitude-level improvements in computational speed and memory efficiency over existing equivariant models while enabling high-precision long-time simulations of organic systems containing hundreds of thousands of atoms using only 901 samples from a 120-atom dataset.
Key Contributions
- A universal multiscale higher-order equivariant model integrated with active learning techniques is developed to efficiently capture complex long-range intermolecular interactions and molecular conformations in large-scale organic systems.
- A bond length stretching method is introduced to enhance numerical stability during extended molecular dynamics simulations.
- The approach achieves superior predictive accuracy with magnitude-level improvements in computational speed and memory efficiency, scaling to systems containing hundreds of thousands of atoms using only 901 training samples from a 120-atom dataset.
Introduction
Molecular simulations are essential for advancing organic material science, but traditional ab initio methods are prohibitively expensive while empirical force fields lack sufficient accuracy. Machine learning force fields bridge this gap, yet their deployment in large-scale organic systems remains difficult due to complex long-range interactions, diverse molecular conformations, and instability during extended simulations. To address these bottlenecks, the authors leverage a universal multiscale higher-order equivariant model combined with active learning to efficiently capture intricate intermolecular dynamics. This architecture delivers superior predictive accuracy alongside significant gains in computational speed and memory efficiency. The researchers also introduce a bond length stretching technique to maintain numerical stability over long timeframes, enabling high-precision simulations of systems with hundreds of thousands of atoms using only 901 training samples.
Dataset

-
Dataset Composition and Sources
- The authors generate data for perfluorotri-n-butylamine (C12F27N), a dielectric liquid coolant, using molecular dynamics simulations.
- Initial conformations are sampled from a 60 ns annealing trajectory cooled from 800 K to 280 K using the GAFF force field. Three hundred frames are extracted at 10 ps intervals.
- Additional high-temperature configurations are gathered via MLFF-guided MD simulations at 300 K, 500 K, 700 K, and 900 K.
- All energy and force labels are computed using the semi-empirical GFN2-xTB method to balance computational cost and accuracy.
-
Key Details for Each Subset
- The final training set contains 901 conformations, with each sample consisting of three molecules (120 atoms) selected from simulation frames.
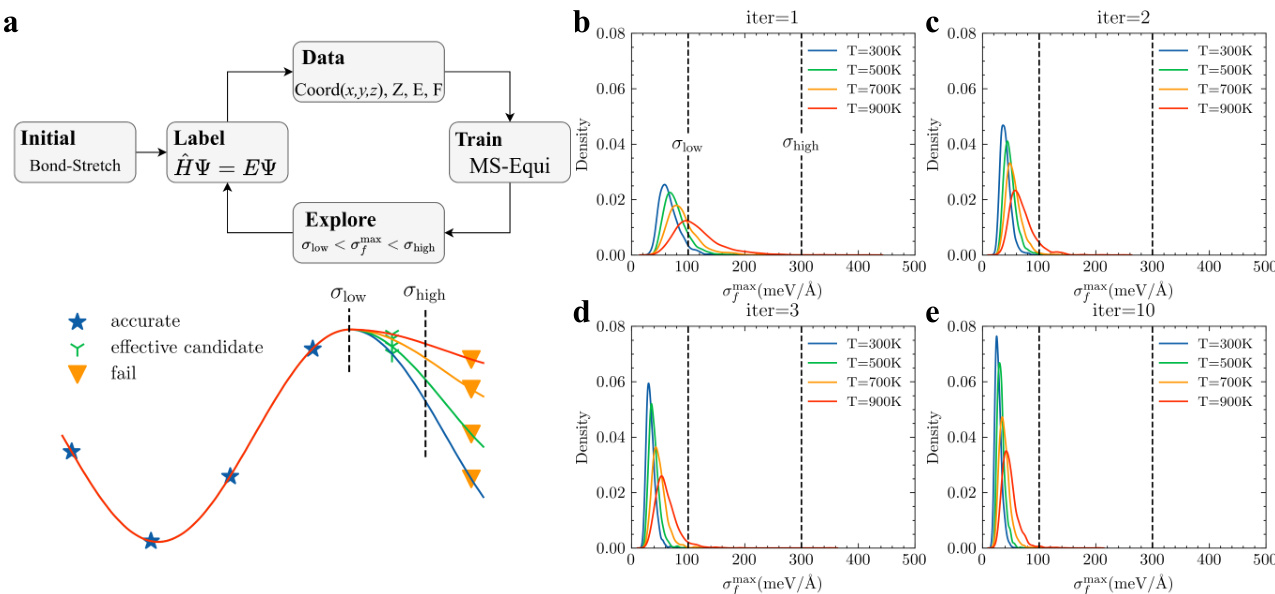
- An active learning pipeline filters candidates based on maximum force variance across a committee of four models trained with different random seeds.
- Selection relies on two thresholds: a lower limit of 100 meV/Å and an upper limit of 300 meV/Å.
- Conformations with variance below the lower threshold are considered redundant and excluded. Samples exceeding the upper threshold are flagged as abnormal and discarded.
- Only candidates falling within the target range are retained, and data collection halts once the effective candidate rate drops below 0.5%.
-
Data Usage and Training Setup
- The curated dataset trains multiscale equivariant force field models, specifically MS-MACE and MS-NequIP.
- Model performance is evaluated on a separate test set by measuring mean absolute errors for energy and forces across varying bond lengths.
- Long-time simulation stability is verified through 50,000-step Langevin dynamics runs at 800 K, with collapse defined as any bond deviating more than 5 Å from equilibrium.
- The authors compare models trained on the original 300-sample subset against an augmented variant, though no fixed train-validation-test split is explicitly detailed.
-
Processing and Augmentation Strategies
- To improve generalization at extreme bond lengths, the authors apply a bond stretching augmentation to the initial 300-sample subset.
- Each conformation undergoes random bond multiplication by a factor drawn uniformly from 0.85 to 2.0, creating an expanded variant for comparative training.
- Spatial cropping is handled by selecting the three molecules closest to the simulation box center from each extracted frame.
- All quantum mechanical labels and variance metrics are computed consistently using GFN2-xTB defaults to ensure uniform metadata across the dataset.
Method
The authors leverage a universal multiscale higher-order equivariant modeling framework designed to efficiently capture both short-range and long-range intermolecular interactions in molecular systems. The overall architecture, as illustrated in the framework diagram, consists of three primary components: an embedding layer, an interaction layer, and an output layer. The interaction layer is composed of multiple sequential blocks, each responsible for processing interactions between a central atom and its neighboring atoms. The model employs a dual-path strategy to handle interactions at different spatial scales. A high-cost module is used for short-range interactions, capturing detailed local features, while a low-cost module handles long-range intermolecular interactions. This multiscale approach enables the model to achieve optimal precision and efficiency in modeling systems with significant long-range effects.
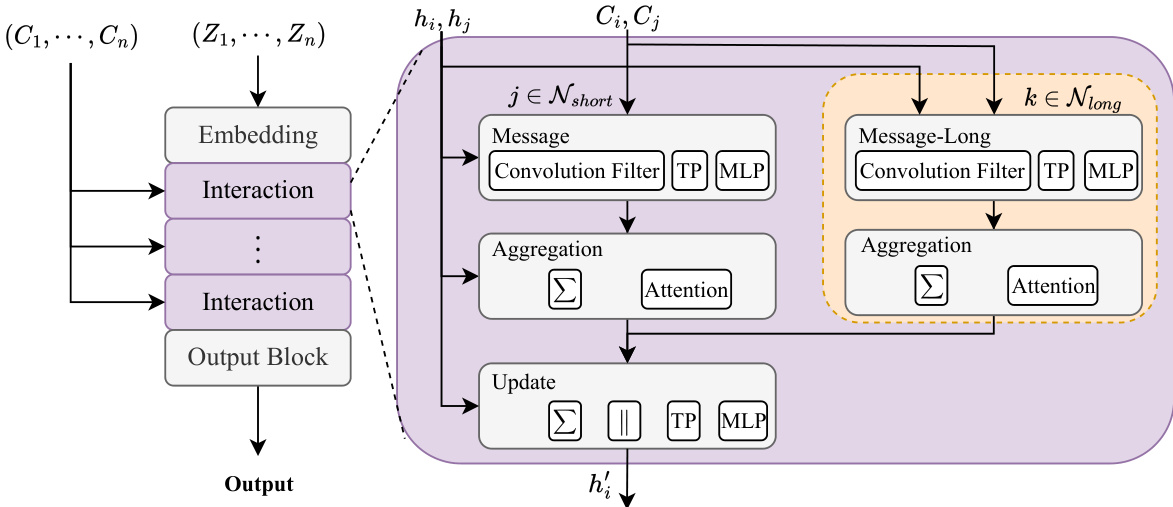
The model is built upon the MACE framework, which is a higher-order equivariant model. The process begins with the atomic numbers Zi being mapped to a one-dimensional vector δzi through one-hot encoding. A linear transformation then initializes the node features hi,c00(0), which represent scalar information. In subsequent interaction layers, node features are denoted as hˉi,cl2m2(t,short/long), where t represents the interaction layer, and "short/long" distinguishes between the two interaction paths. The key design choice is that the short-range path uses a larger number of channels and a higher-order expansion for directional information, while the long-range path uses a smaller number of channels and a lower-order expansion, significantly reducing computational complexity. The framework diagram shows that these two paths operate in parallel, with the long-range path being processed within an orange-dashed box.
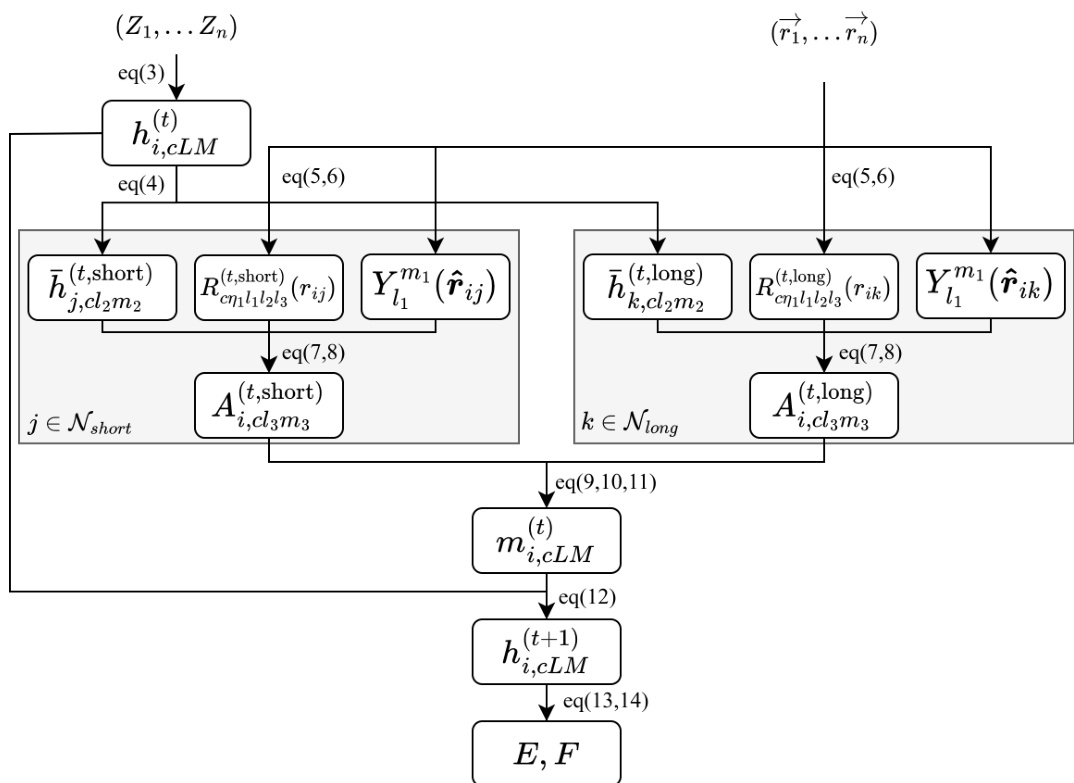
The interaction layer processes the features through a series of steps for both short- and long-range interactions. The initial step is a linear transformation that satisfies equivariance requirements, ensuring that the transformation preserves the symmetry properties of the data. This is followed by radial embedding, where the interatomic distance rij is transformed using Bessel basis functions and a polynomial smooth truncation function. The radial information is then expanded to a specified dimension using a learnable multilayer perceptron (MLP). The core of the interaction is a convolutional filtering operation, where the radial and directional information are combined with neighboring atomic features to produce a message. This message is aggregated and then updated through another MLP and a residual connection, resulting in the updated node features for the next layer.
The final potential energy Epot is computed as the sum of the contributions from all atoms, where each atom's energy is the sum of its short-range and long-range components. The atomic forces are derived as the negative gradient of the total potential energy with respect to the atomic coordinates, ensuring energy conservation. The entire process is designed to be efficient, with the long-range module using a smaller channel dimension and a lower-order expansion to reduce computational cost. The calculation flow diagram illustrates that a single interaction layer transforms the node features from hi,cLM(t) to hi,cLM(t+1), and the final energy is obtained by summing the linearly read-out results from each layer's features. The forces are then computed from the gradient of the final energy.
Experiment
The evaluation compares the proposed multiscale equivariant model against established architectures and ab initio molecular dynamics simulations across systems scaling from hundreds to hundreds of thousands of atoms. The benchmarking experiments validate predictive accuracy and computational efficiency against competing equivariant models, while structural validation experiments confirm fidelity against quantum mechanical baselines. Qualitatively, the results demonstrate that the proposed model consistently outperforms existing methods by maintaining high prediction precision at large scales while achieving substantial improvements in simulation speed and memory efficiency. Overall, the findings confirm that the multiscale approach successfully extends accurate and scalable molecular dynamics simulations to large-scale systems without performance degradation.
The authors compare their multiscale model with other equivariant models in terms of prediction accuracy, simulation speed, and GPU memory consumption. Results show that their model achieves the lowest prediction errors and significantly better efficiency across all metrics, with linear scaling of time and memory costs as the number of atoms increases. The proposed model achieves the lowest prediction errors for both energy and forces compared to other models. The model demonstrates superior computational efficiency with significantly lower simulation time and GPU memory usage. All models exhibit linear scaling of time and memory consumption with increasing atomic number, indicating consistent performance on larger systems.
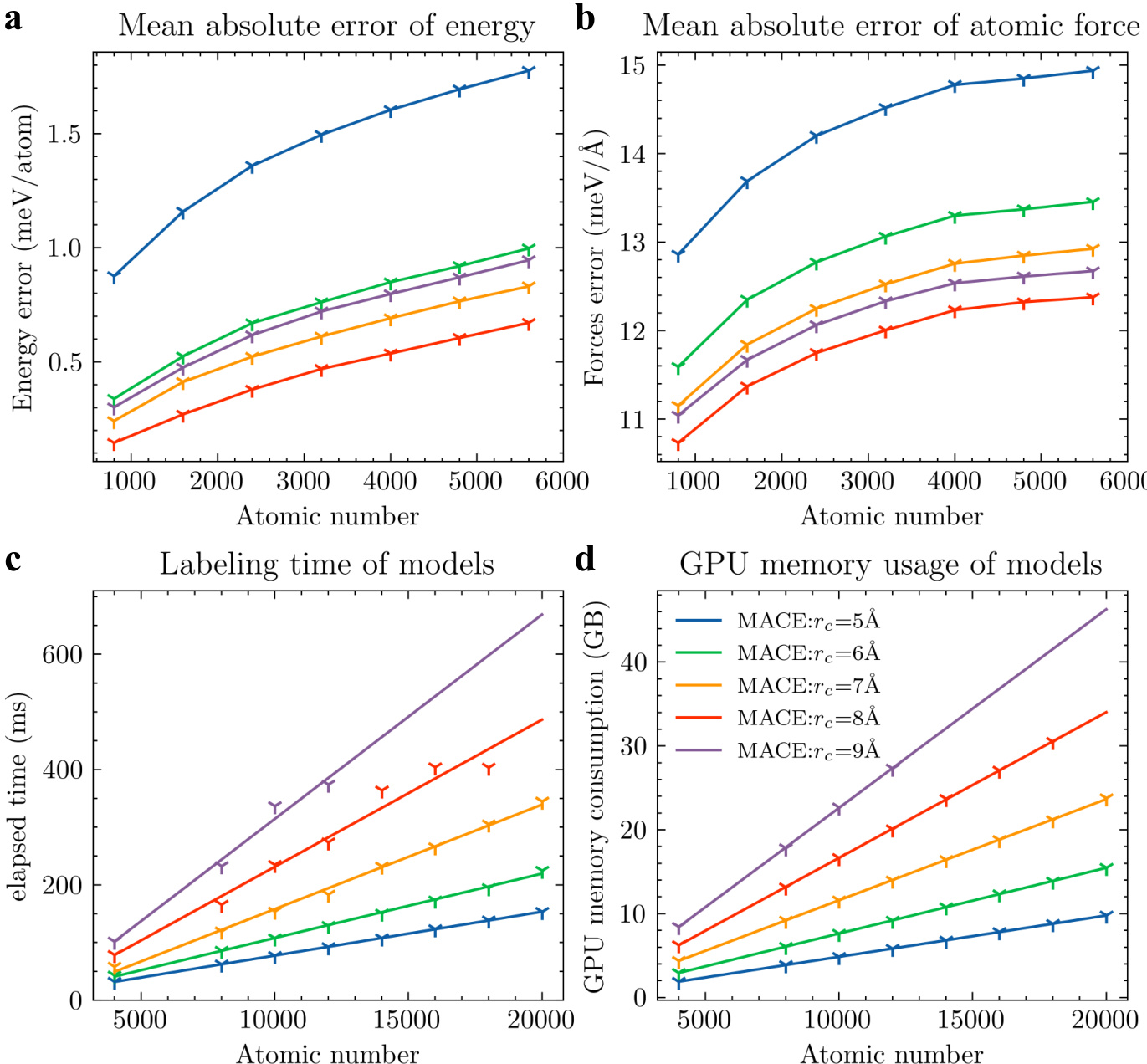
The authors compare their multiscale model with other equivariant models in terms of prediction accuracy, simulation speed, and GPU memory consumption. Results show that their model achieves the lowest error in energy and force predictions, and demonstrates superior computational efficiency and memory usage across varying system sizes. The performance improvements become more pronounced as the number of atoms increases. The proposed model achieves the lowest prediction errors for both energy and forces compared to other models. The model exhibits significantly better computational efficiency and memory consumption, with improvements becoming more evident at larger system sizes. The performance gains are consistent across different system sizes, indicating scalability and robustness.
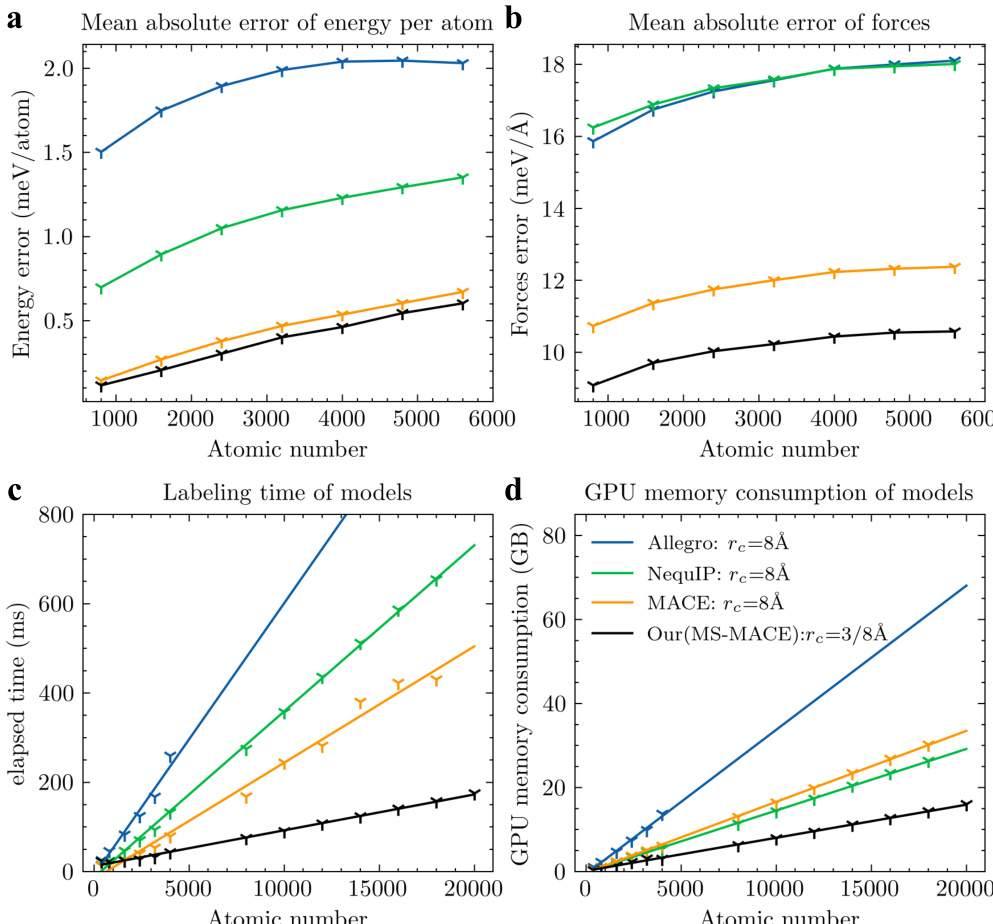
The authors compare their multiscale model with other equivariant models in terms of prediction accuracy, simulation speed, and GPU memory consumption. The results show that their model achieves the best performance across all metrics, with high accuracy and significant improvements in computational efficiency. The model maintains consistent performance as the system size increases, demonstrating its scalability. The proposed model outperforms other equivariant models in accuracy, simulation speed, and GPU memory consumption. The model maintains high prediction accuracy even as the system size increases, indicating scalability. The model achieves significant improvements in computational efficiency, with linear scaling of time cost and memory consumption.
The experiments benchmark the proposed multiscale model against established equivariant architectures by evaluating prediction accuracy, simulation speed, and memory efficiency across systems of increasing complexity. The validation demonstrates that the new framework consistently yields more accurate energy and force predictions while requiring substantially less computational time and GPU resources. These efficiency gains scale linearly with system size, confirming that the model maintains robust performance without sacrificing accuracy on larger datasets. Overall, the results establish the multiscale approach as a highly scalable and computationally superior alternative to existing methods.