Command Palette
Search for a command to run...
使用 VASP 进行机器学习力场训练
摘要
一句话总结
作者提出了一种集成主动学习与键长拉伸方法的全能多尺度高阶等变机器学习力场。该力场在预测精度上达到最高水平,并在计算速度与内存效率上较现有等变模型实现量级提升,同时仅使用来自120原子数据集的901个样本,即可对包含数十万原子的有机体系进行高精度长时间模拟。
核心贡献
- 开发了一种集成主动学习技术的全能多尺度高阶等变模型,以高效捕捉大规模有机体系中的复杂长程分子间相互作用与分子构象。
- 引入键长拉伸方法,以提升长时间分子动力学模拟过程中的数值稳定性。
- 该方法实现了卓越的预测精度,计算速度与内存效率实现量级提升,仅需120原子数据集的901个训练样本,即可扩展至包含数十万原子的体系。
引言
分子模拟对于推进有机材料科学至关重要,但传统的从头算方法成本过高,而经验力场又缺乏足够的精度。机器学习力场填补了这一空白,但由于复杂的长程相互作用、多样的分子构象以及长时间模拟中的不稳定性,其在大规模有机体系中的部署仍面临挑战。为解决这些瓶颈,作者结合主动学习技术,利用全能多尺度高阶等变模型来高效捕捉复杂的分子间动力学过程。该架构在提供卓越预测精度的同时,大幅提升了计算速度与内存效率。研究人员还引入了键长拉伸技术以维持长时间范围内的数值稳定性,使得仅凭901个训练样本即可对包含数十万原子的体系进行高精度模拟。
数据集
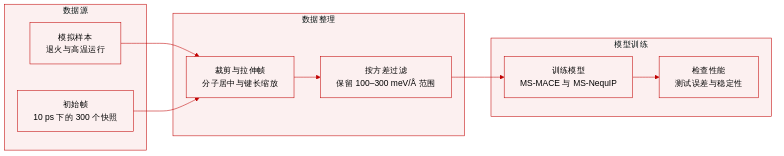
-
数据集构成与来源
- 作者通过分子动力学模拟生成了全氟三正丁胺(C12F27N,一种介电液体冷却剂)的数据。
- 初始构象从使用GAFF力场从800 K冷却至280 K的60纳秒退火轨迹中采样。每隔10皮秒提取一帧,共提取300帧。
- 额外的高温构象通过机器学习力场(MLFF)引导的分子动力学模拟在300 K、500 K、700 K和900 K下收集。
- 所有能量与力标签均采用半经验GFN2-xTB方法计算,以平衡计算成本与精度。
-
各子集关键细节
- 最终训练集包含901个构象,每个样本由从模拟帧中选出的三个分子(共120个原子)组成。
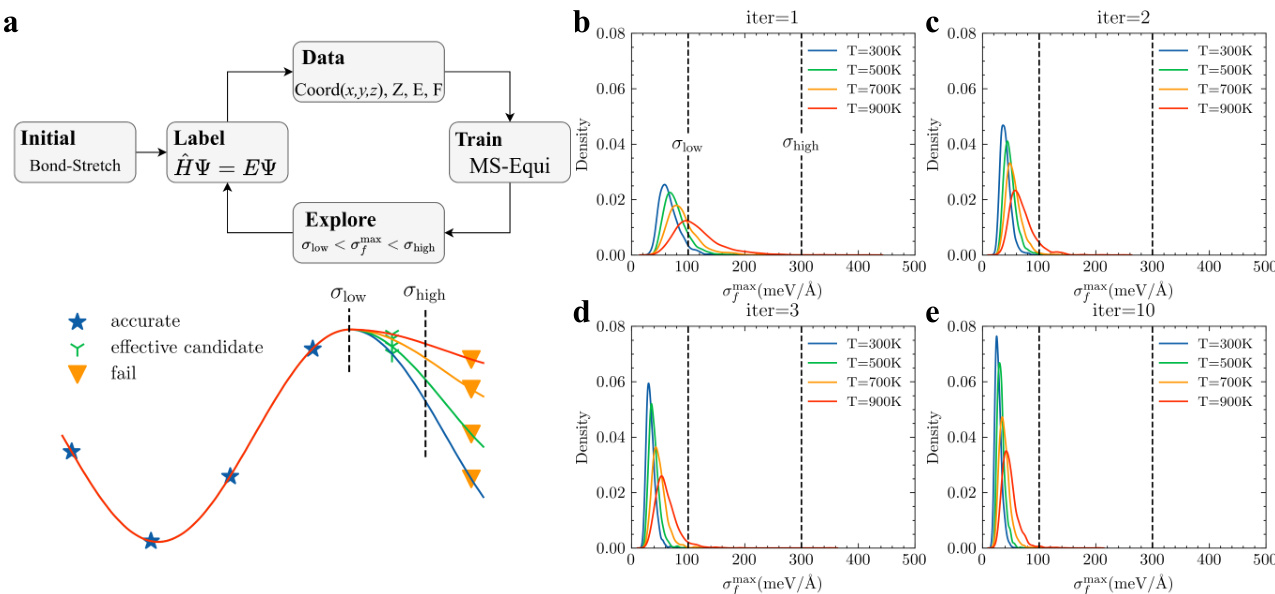
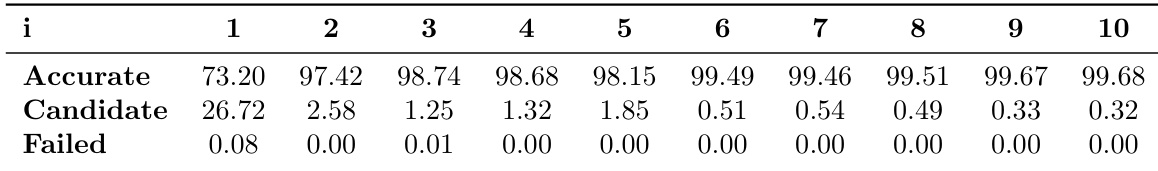
- 主动学习流水线基于由四个使用不同随机种子训练的模型组成的委员会的最大力方差来筛选候选样本。
- 筛选依赖两个阈值:下限为100 meV/Å,上限为300 meV/Å。
- 方差低于下限的构象被视为冗余并予以排除。超过上限的样本被标记为异常并丢弃。
- 仅保留落在目标范围内的候选样本,当有效候选率降至0.5%以下时,数据收集即告停止。
-
数据使用与训练设置
- 经筛选的数据集用于训练多尺度等威力场模型,具体包括MS-MACE与MS-NequIP。
- 模型性能通过在独立测试集上测量不同键长下的能量与力的平均绝对误差进行评估。
- 通过800 K下50,000步的朗之万动力学运行验证长时间模拟的稳定性,其中任意键偏离平衡位置超过5 Å即定义为崩溃。
- 作者对比了在原始300样本子集与增强变体上训练的模型,但未明确说明固定的训练集、验证集与测试集划分比例。
-
处理与增强策略
- 为提升极端键长下的泛化能力,作者对初始300样本子集应用了键拉伸增强。
- 每个构象的键长均乘以从0.85至2.0均匀分布中抽取的随机因子,从而生成用于对比训练的扩展变体。
- 空间裁剪通过从每帧提取的构象中选择距离模拟盒子中心最近的三个分子来实现。
- 所有量子力学标签与方差指标均统一使用GFN2-xTB默认参数计算,以确保数据集元数据的一致性。
方法
作者采用了一种全能多尺度高阶等变建模范式,旨在高效捕捉分子体系中的短程与长程分子间相互作用。如图文框架所示,整体架构包含三个核心部分:嵌入层、交互层与输出层。交互层由多个顺序排列的模块组成,每个模块负责处理中心原子与其邻近原子之间的相互作用。该模型采用双路径策略来处理不同空间尺度的相互作用。高计算成本模块用于短程相互作用,以捕捉详细的局部特征;低计算成本模块则负责长程分子间相互作用。这种多尺度方法使模型能够在建模具有显著长程效应的体系时,实现精度与效率的最优平衡。
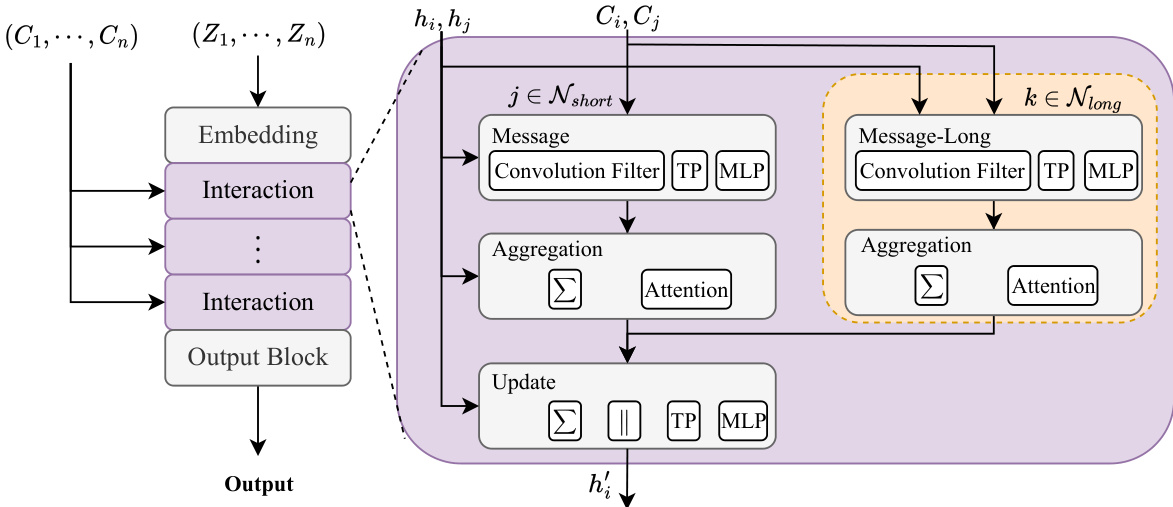
该模型基于MACE框架构建,MACE是一种高阶等变模型。处理首先将原子序数 Zi 通过独热编码映射为一维向量 δzi。随后通过线性变换初始化表示标量信息的节点特征 hi,c00(0)。在后续的交互层中,节点特征记为 hˉi,cl2m2(t,short/long),其中 t 代表交互层索引,"short/long" 用于区分两条交互路径。关键设计在于:短程路径采用更多的通道数与高阶展开来处理方向信息,而长程路径则使用较少的通道数与低阶展开,从而大幅降低计算复杂度。框架图显示这两条路径并行运行,长程路径的处理过程包含在橙色虚线框内。
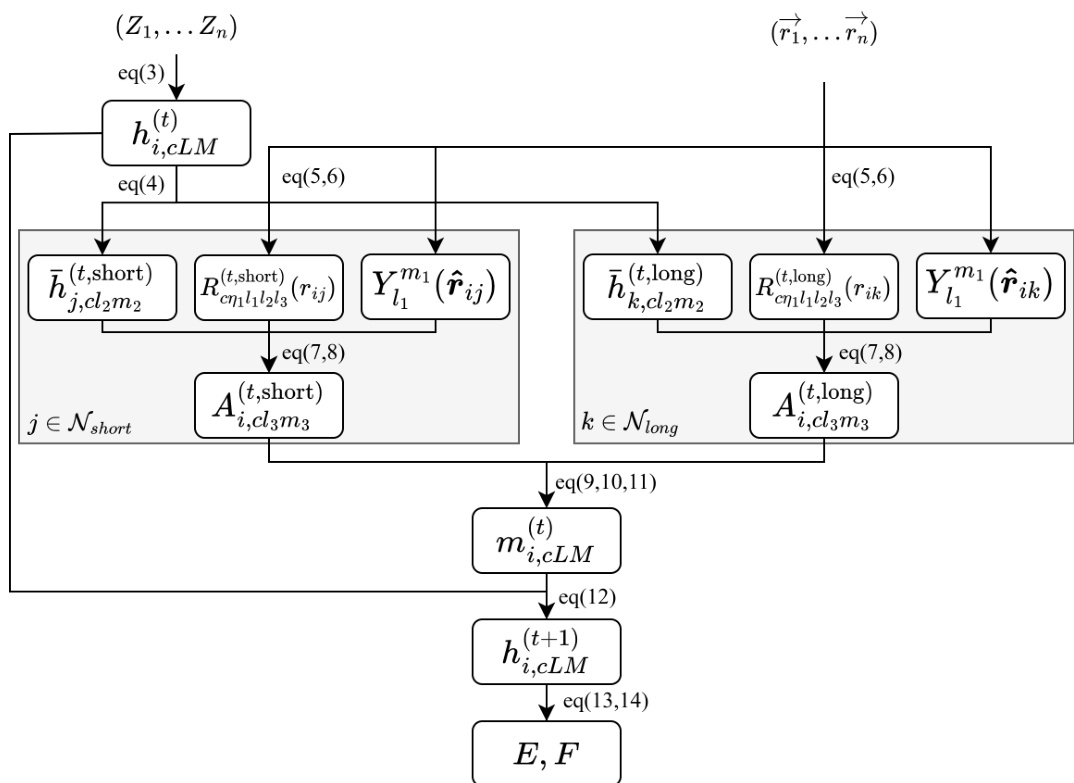
交互层通过一系列步骤处理短程与长程相互作用的特征。初始步骤为满足等变性要求的线性变换,确保变换过程保留数据的对称性特征。随后进行径向嵌入,利用贝塞尔基函数与多项式平滑截断函数对原子间距离 rij 进行变换。径向信息随后通过可学习的多层感知机(MLP)扩展至指定维度。交互的核心是卷积滤波操作,径向与方向信息在此与邻近原子特征结合以生成消息。该消息经过聚合后,通过另一个MLP与残差连接进行更新,从而得到下一层的更新节点特征。
最终势能 Epot 计算为所有原子贡献之和,其中每个原子的能量为其短程与长程分量之和。原子力通过总势能关于原子坐标的负梯度求导获得,从而保证能量守恒。整个过程以高效为目标设计,长程模块采用较小的通道维度与低阶展开以降低计算成本。计算流程图显示,单个交互层将节点特征从 hi,cLM(t) 变换为 hi,cLM(t+1),最终能量通过对各层特征的线性读出结果求和获得。随后根据最终能量的梯度计算原子力。
实验
评估实验将所提出的多尺度等变模型与成熟架构及从头算分子动力学模拟进行对比,测试体系规模涵盖数百至数十万原子。基准测试实验验证了模型在预测精度与计算效率方面相对于其他等变模型的优势,结构验证实验则确认了模型与量子力学基线的一致性。定性结果表明,该模型在大规模体系下始终保持高预测精度,同时在模拟速度与内存效率方面实现显著提升,整体性能稳定优于现有方法。总体而言,研究结果证实多尺度方法成功将高精度且可扩展的分子动力学模拟扩展至大规模体系,且未出现性能衰减。
作者从预测精度、模拟速度与GPU显存消耗三个维度,将所提出的多尺度模型与其他等变模型进行对比。结果表明,该模型在所有指标上均取得最低的预测误差与显著更优的效率,且时间与内存成本随原子数量增加呈线性扩展。与其他模型相比,所提模型在能量与力的预测误差上均达到最低。该模型展现出卓越的计算效率,模拟时间与GPU显存占用显著降低。所有模型的时间与内存消耗均随原子数量增加呈线性扩展,表明其在更大体系上性能保持一致。
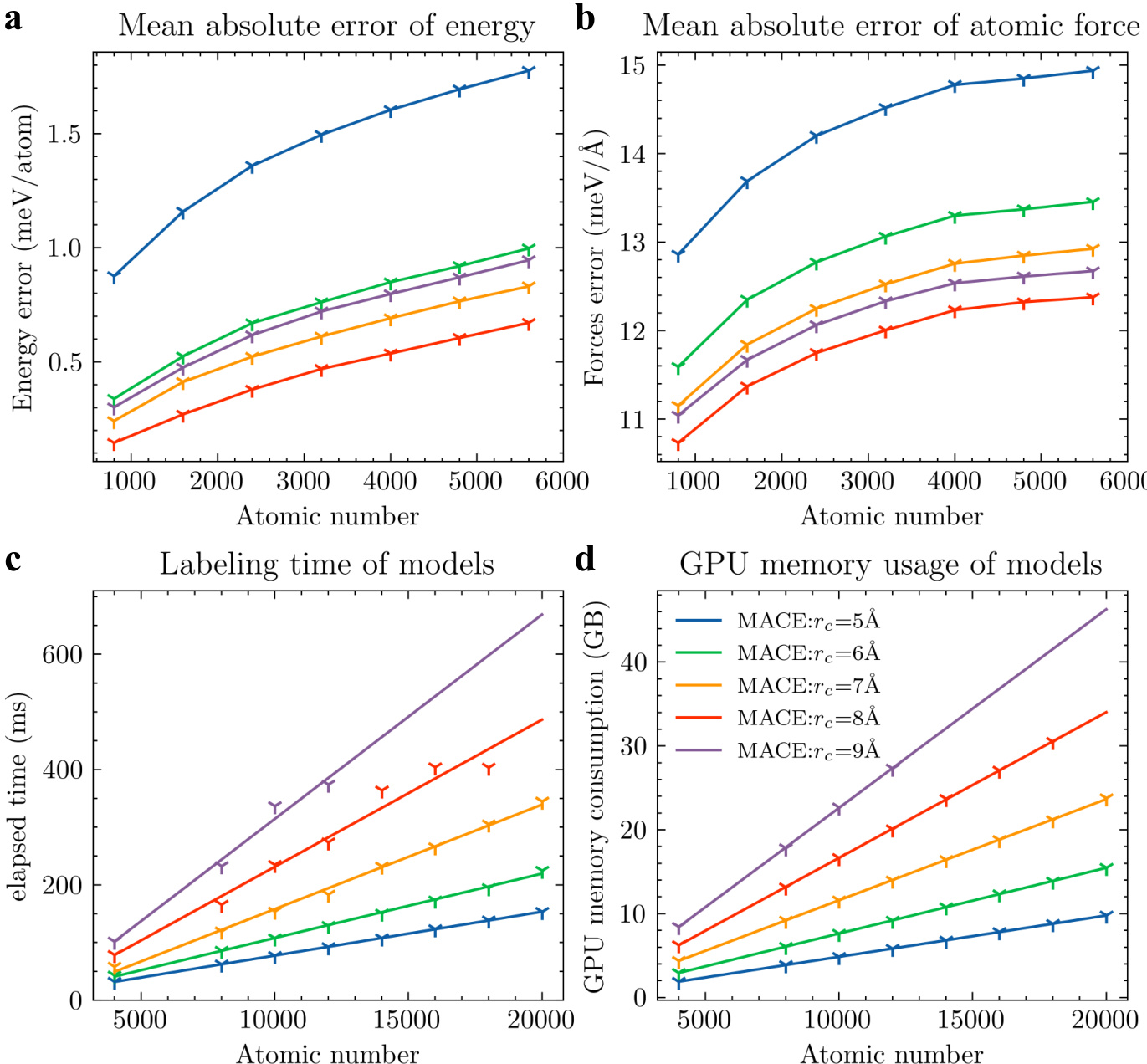
作者从预测精度、模拟速度与GPU显存消耗三个维度,将所提出的多尺度模型与其他等变模型进行对比。结果表明,该模型在能量与力预测误差上最低,并在不同体系规模下展现出更优的计算效率与内存使用表现。随着原子数量增加,性能提升效果更为显著。与其他模型相比,所提模型在能量与力的预测误差上均达到最低。该模型在计算效率与内存消耗方面表现显著更优,且规模越大优势越明显。性能增益在不同体系规模下保持一致,表明该模型具备良好的可扩展性与鲁棒性。
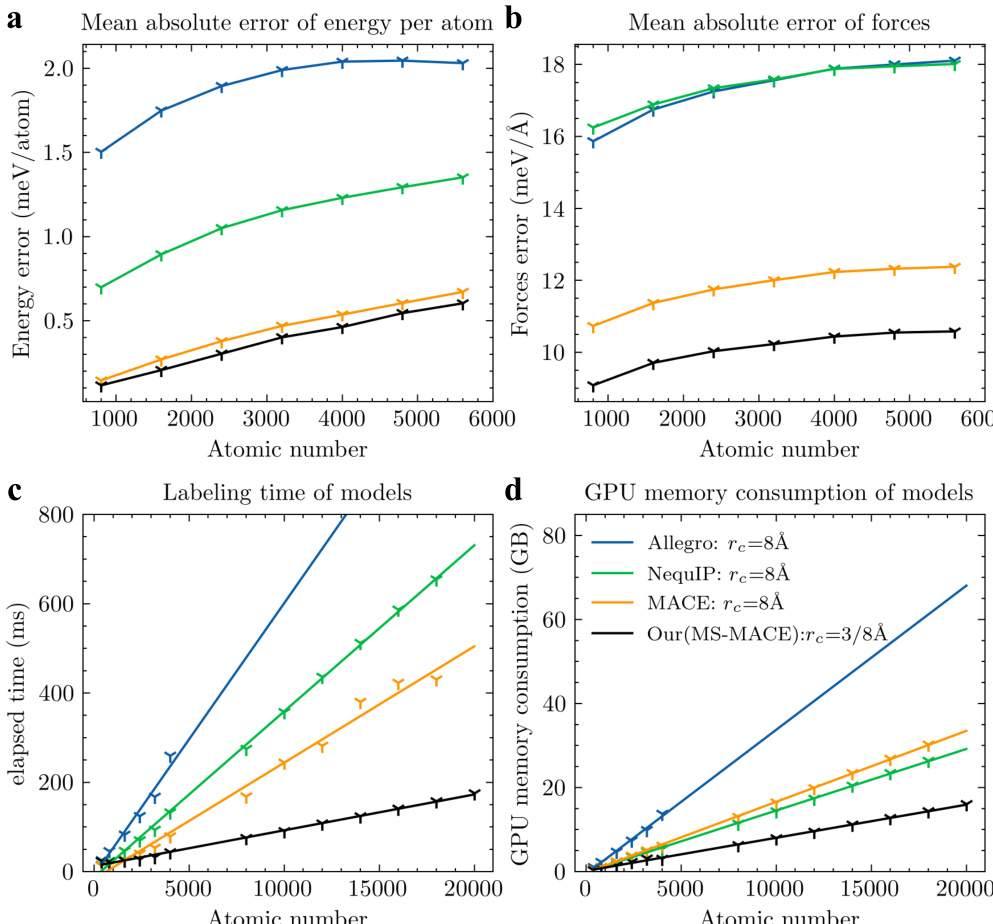
作者从预测精度、模拟速度与GPU显存消耗三个维度,将所提出的多尺度模型与其他等变模型进行对比。结果表明,该模型在所有指标上均取得最佳性能,兼具高精度与计算效率的显著提升。随着体系规模扩大,模型性能保持稳定,展现出良好的可扩展性。所提模型在精度、模拟速度与GPU显存消耗方面均优于其他等变模型。即使体系规模扩大,模型仍能维持高预测精度,进一步印证其可扩展性。该模型在计算效率方面实现显著提升,时间与内存成本呈线性扩展。
实验通过在复杂度递增的体系上评估预测精度、模拟速度与内存效率,将所提出的多尺度模型与成熟等变架构进行基准对比。验证结果表明,新框架始终能输出更精确的能量与力预测,同时大幅降低计算时间与GPU资源需求。这些效率增益随体系规模呈线性扩展,证实该模型在更大规模数据上仍能保持稳健性能且不牺牲精度。总体而言,结果确立了多尺度方法作为一种高度可扩展且计算性能显著优于现有方法的替代方案。